Prof Jonathan Essex
- Position
- Professor
- Institution
- Chemistry (FNES)
- Webpage
- http://www.soton.ac.uk/~chemphys/jessex/group.html
- Contact
- Complete this online contact form to contact Jonathan.
Research
Computer simulations of molecular systems are a vital component of modern chemistry and physics. They are being used, for example, in such diverse areas of research as the fundamental physics of crystal nucleation, through to the design of new pharmaceutical entities. Research in the Essex group focuses on innovation in the application of computer simulations to biological systems, where there is the potential to contribute to drug discovery and the development of medical diagnostics. A key challenge restricting success has been limitations in the range of applicability of these computational methods, and in the extent to which accurate predictions may be made. To address these issues, a cross-disciplinary approach that develops new methodologies and deploys these over more realistic systems is actively sought.
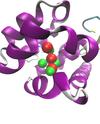
Protein-ligand interactions are critical in determining the effectiveness of pharmaceutical compounds, and computer simulations are ideally suited to probing these interactions. There are essentially two problems to be solved: First, is it possible to predict the structure of a protein-ligand complex given the protein structure, and second, can the binding free energy of the resulting complex be accurately calculated? If these objectives can be met, then the reliable use of structure to predict novel pharmaceutical compounds will become a reality.
Past work involved the developement of structure prediction algorithms that incorporates receptor flexibility. This method is currently undergoing further testing and validation. The role of water molecules present in a protein ligand binding site is the subject of much debate and developing computational methods that consider their role for ligand design is a challenging task. The binding free energy of these water molecules can be calculated by application of the formalism of statistical thermodynamics. This permit to discriminate between water molecules that can be displaced by ligands and those that are tightly bound and should not be targeted.
Reliable binding free energy calculations requires extensive sampling of the range of possible ligand-protein interactions to yield meaningful results. Protocols that combine Replica Exchange methodology (used extensively in molecular dynamics simulation of protein conformational changes) and free energy calculations have been developed in the past. Current work considers non equilibrium methods, novel Monte Carlo moves and approximate methods of solvation to improve the efficiency of these computations. Group members working in this field :
- Frank Beierlein: IntBioSim: An Integrated Approach to Multi-Level Biomolecular Simulations
- Patrick Schöpf: The Development and application of free-energy calculations in rational drug-design.
- Genevieve Clapton: How kinase point mutations confer resistance: a computational study
- Nadia Vahdati: The modelling of receptor flexibility on ligand binding
- Juan Fernandez-Carmona: Clever moves for protein backbone using the MC method
- Michael Bodnarchuk: The Use of Molecular Fragments in Inhibitor Design
Biological membranes
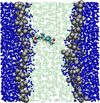
Computer simulations of biological systems have, until recently, been limited to at most 100 000 atoms. This is sufficient to study a single large protein in an aqueous environment. However, real biological systems are substantially larger than this, and if simulation is ever to offer insight into how, for example, lipids and proteins interact in a fully functioning cell membrane, approaches to simplify the atomic representation and increase the granularity of our models are needed.
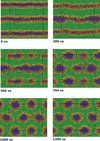
Past work has focused on atomistic simulations of small molecule and drug permeation through model membranes. These simulations have provided insights in understanding how readily drugs reach their target. Simplified molecular models have long been used in the area of modelling liquid crystal systems, with considerable success. A simplified model of the membrane environment, based on the Gay-Berne model of liquid crystals has been developed.
Future work will likely focus on coarse-grained models of protein and DNA. These studies will address not only the fundamental physics needed to develop successful coarse-grained models, but will also attack important biological problems such as protein-mediated membrane fusion. Group members working in this field :
- Mario Orsi: The Simulation of Coarse-Grained Biomembranes and Drug Transport therein via Anisotropic Molecular Dynamics Models
- George Chellapa: The development of simplified lipid models using a Molecular Dynamics Coarse Graining approach
Protein Conformational Change
One of the major limitations of conventional atom-based computer simulations is the inability to simulate long enough to study rare events, such as conformational change in proteins. This is often an integral part of the proteins function, requiring times in excess of microseconds to be observed. The fundamental limitations of conventional simulation methodology, are being adressed by the application of a novel technique to accelerate simulations and access these rare motions.
The application of digital signal processing to the molecular dynamics simulation velocities allows the atomic motion to be enhanced or suppressed in a selective manner solely on the basis of vibrational frequency. This methodology has been successfully applied to the simulation of the conformational changes in lysozyme, dihydrofolate reductase and HIV-protease. The analysis of these simulations is being assisted by the application of the recently developed Hilbert-Huang Transform, to allow the amplitude of molecular vibrations over defined frequency ranges to be calculated as a function of time.
Currently we are looking at Hybrid molecular dynamics / Monte Carlo and Replica Exchange simulation methods as alternative/complementary means to observe protein conformational changes. These methodologies are also applied to study the conformational equilibria in kinases. These enzymes are a very important drug target for the treatment of cancer, showing significant conformational change, and indeed the development of drug resistant mutations in these proteins is proving increasingly worrisome.
Group members working in this field :
- Nadia Vahdati: The modelling of receptor flexibility on ligand binding
- Mishal Patel: The development and application of simplified protein models
- Ifat Noreen: The development and implementation of hybrid Molecular Dynamics/Monte Carlo methods
- Juan Fernandez-Carmona: Clever moves for protein backbone using the MC method
Jonathan's team members
 Michael Bodnarchuk Michael BodnarchukPostgraduate Research Student, Chemistry (FNES) |
|
James Graham Postgraduate Research Student, Electronics and Computer Science (FPAS) |
 Sophia Wheeler Sophia WheelerPostgraduate Research Student, Electronics and Computer Science (FPAS) |
 Kieran Selvon Kieran SelvonPostgraduate Research Student, Engineering Sciences (FEE) |
 Petrina Butler Petrina ButlerAdministrative Staff, Research and Innovation Services |
|
Barbara Sander Alumnus, Chemistry (FNES) |
 Dan Mason Dan MasonAlumnus, University of Southampton |
Joint projects with...
 Seth Bullock Seth BullockProfessor, Electronics and Computer Science (FPAS) |
 Hans Fangohr Hans FangohrProfessor, Engineering Sciences (FEE) |
 Chris-Kriton Skylaris Chris-Kriton SkylarisLecturer, Chemistry (FNES) |
Research Groups
Head of Institute for Complex Systems Simulations (ICSS)
University of Southampton
Projects

Complex Systems Simulations Centre for Doctoral Training
With Seth Bullock, Hans Fangohr (Investigators)

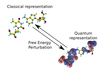
Hybrid quantum and classical free energy methods in computational drug optimisation
With Chris-Kriton Skylaris (Investigator), Christopher Cave-Ayland