Onetep
ONETEP is a linear-scaling density functional theory software package able to run on parallel computers[1]. It uses a basis of non-orthogonal generalized Wannier functions (NGWFs) expressed in terms of periodic cardinal sine (psinc) functions, which are in turn equivalent to a basis of plane-waves. ONETEP therefore combines the advantages of the plane-wave approach (controllable accuracy and variational convergence of the total energy with respect to the size of the basis) with computational effort that scales linearly with the size of the system [2]. The ONETEP approach involves simultaneous optimization of the density kernel (a generalization of occupation numbers to non-orthogonal basis, which represents the density matrix in the basis of NGWFs) and the NGWFs themselves. The optimized NGWFs then provide a minimal localized basis set, which can be considerably smaller than the unoptimized basis sets used in most approaches.
ONETEP has been developed by a group of UK academics based at Cambridge University, Southampton University and Imperial College London. It is available free to UK academics, and licenses can be obtained for non-UK academic usage from the developers or through Accelrys' Materials Studio package.
Official website: http://www2.tcm.phy.cam.ac.uk/onetep/Main/HomePage Wikipedia entry: http://en.wikipedia.org/wiki/ONETEP
For queries about this topic, contact Chris-Kriton Skylaris.
View the calendar of events relating to this topic.
Projects

Ab initio simulations of chemical reactions on platinum nanoparticles
Chris-Kriton Skylaris (Investigator), Álvaro Ruiz-Serrano, Peter Cherry
•Use first principles calculations to study the relationship between shape and size of nanoparticle and the oxygen adsorption energy.
• Investigate the effect of high oxygen coverage on the catalytic activity of the nanoparticles.
Can we calculate the pKa of new drugs, based on their structure alone?
Chris-Kriton Skylaris (Investigator), Chris Pittock, Jacek Dziedzic
The pKa of an active compound in a pharmaceutical drug affects how it is absorbed and distributed around the human body. While there are various computational methods to predict pKa using only molecular structure data, these tend to be specialised to only one class of drug - we aim to generate a more generalised prediction method using quantum mechanics.

Centre for Doctoral Training in Next Generation Computational Modelling
Hans Fangohr, Ian Hawke, Peter Horak (Investigators), Susanne Ufermann Fangohr, Thorsten Wittemeier, Kieran Selvon, Alvaro Perez-Diaz, David Lusher, Ashley Setter, Emanuele Zappia, Hossam Ragheb, Ryan Pepper, Stephen Gow, Jan Kamenik, Paul Chambers, Robert Entwistle, Rory Brown, Joshua Greenhalgh, James Harrison, Jonathon Waters, Ioannis Begleris, Craig Rafter
The £10million Centre for Doctoral Training was launched in November 2013 and is jointly funded by EPSRC, the University of Southampton, and its partners.
The NGCM brings together world-class simulation modelling research activities from across the University of Southampton and hosts a 4-year doctoral training programme that is the first of its kind in the UK.
Development of wide-ranging functionality in ONETEP
Chris-Kriton Skylaris (Investigator), Jacek Dziedzic
ONETEP is at the cutting edge of developments in first principles calculations. However, while the fundamental difficulties of performing accurate first-principles calculations with linear-scaling cost have been solved, only a small core of functionality is currently available in ONETEP which prevents its wide application. In this collaborative project between three Universities, the original developers of ONETEP will lead an ambitious workplan whereby the functionality of the code will be rapidly and significantly enriched.
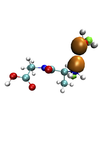
Dipole moment and theoretical spectroscopy: a computational approach
Chris-Kriton Skylaris (Investigator), Valerio Vitale
The present project represents a first step towards the implementation of a new technique to calculate the whole vibrational spectra of molecules in a formally exact way, which fully takes into account anharmonicity and conformational transitions, at a finite temperature, both in gas phase and in solution in a single ab initio molecular dynamics simulation.
Electrostatic embedded energy calculations of proteins, using the ONETEP DFT code
Chris-Kriton Skylaris (Investigator), Stephen Fox, Chris Pittock
Calculating the energy of a biomolecule in solvent, using quantum mechanics (QM) is possible, but extremely challenging, even with linear-scaling QM methods like ONETEP. Using electrostatic embedding, a novel twist on the existing QM/MM method is used to calculate the binding energy of a small ligand to a solvated protein, increasing the accuracy and realism of our general project work.

Investigation into the Interfacial Physics of Field Effect Biosensors
Nicolas Green, Chris-Kriton Skylaris (Investigators), Benjamin Lowe
This interdisciplinary research aims to improve understanding of Field Effect Transistor Biosensors (Bio-FETs) and to work towards a multiscale model which can be used to better understand and predict device response.

Large-Scale Quantum Chemistry Simulations of Organic Photovoltaics
Chris-Kriton Skylaris (Investigator), Gabriele Boschetto
The aim of this project is to use first principles quantum mechanical calculations to provide a detailed atomic-level understanding of OPV materials and models of bulk heterojunctions on a far larger scale than possible before by using the ONETEP program for linear-scaling first principles quantum mechanical calculations.
Sustainable domain-specific software generation tools for extremely parallel particle-based simulations
Chris-Kriton Skylaris (Investigator)
A range of particle based methods (PBM) are currently used to simulate materials in chemistry, engineering, physics and biophysics. The 4 types of PBM considered directly in the proposed are molecular dynamics (MD), the ONETEP quantum mechanics-based program, discrete element modelling (DEM), and smoothed particle hydrodynamics (SPH).
The overall research objective is to develop a sustainable tool that will deliver, in the future, cutting edge research applicable to applications ranging from dam engineering to atomistic drug design.
The hydrogen abstraction phase of the CYP-cyclohexene reaction, using large-scale DFT
Chris-Kriton Skylaris (Investigator), Chris Pittock, Karl Wilkinson
Studying the hydrogen-abstraction reaction between cyclohexene and the active site of cytochrome P450. This starts a series of reactions that eventually oxidise the small molecule to become either an epoxide or an alcohol.
Understanding the finer detail of this reaction can assist towards a model that will predict the breakdown of drugs in the human body.

Vibrational spectroscopy from ab initio molecular dynamics
Hans Fangohr, Chris-Kriton Skylaris (Investigators), Valerio Vitale
In this project I used the Fourier transform of the time correlation function (FTTCF) formalism, that allows to compute the vibrational spectra of molecules both in gas and condensed phase, at finite temperature, in a single ab initio molecular dynamics simulation.
People
 Hans Fangohr
Hans FangohrProfessor, Engineering Sciences (FEE)
 Nicolas Green
Nicolas GreenReader, Electronics and Computer Science (FPAS)
 Peter Horak
Peter HorakReader, Optoelectronics Research Centre
 Ian Hawke
Ian HawkeLecturer, Mathematics (FSHS)
 Chris-Kriton Skylaris
Chris-Kriton SkylarisLecturer, Chemistry (FNES)
 Jacek Dziedzic
Jacek DziedzicResearch Fellow, Chemistry (FNES)
Karl WilkinsonResearch Fellow, Chemistry (FNES)
Hong-Tao XueResearch Fellow, Chemistry (FNES)
 Ioannis Begleris
Ioannis BeglerisPostgraduate Research Student, Engineering Sciences (FEE)
 Gabriele Boschetto
Gabriele BoschettoPostgraduate Research Student, Engineering Sciences (FEE)
 Rory Brown
Rory BrownPostgraduate Research Student, Civil Engineering & the Environment (FEE)
 Paul Chambers
Paul ChambersPostgraduate Research Student, Engineering Sciences (FEE)
 Peter Cherry
Peter CherryPostgraduate Research Student, Chemistry (FNES)
 Robert Entwistle
Robert EntwistlePostgraduate Research Student, Engineering Sciences (FEE)
Stephen FoxPostgraduate Research Student, Chemistry (FNES)
 Stephen Gow
Stephen GowPostgraduate Research Student, Engineering Sciences (FEE)
 Joshua Greenhalgh
Joshua GreenhalghPostgraduate Research Student, Engineering Sciences (FEE)
 James Harrison
James HarrisonPostgraduate Research Student, Engineering Sciences (FEE)
 Quintin Hill
Quintin HillPostgraduate Research Student, Chemistry (FNES)
 Benjamin Lowe
Benjamin LowePostgraduate Research Student, Electronics and Computer Science (FPAS)
 David Lusher
David LusherPostgraduate Research Student, Engineering Sciences (FEE)
 Alvaro Perez-Diaz
Alvaro Perez-DiazPostgraduate Research Student, Engineering Sciences (FEE)
Maximillian PhippsPostgraduate Research Student, Chemistry (FNES)
 Chris Pittock
Chris PittockPostgraduate Research Student, Chemistry (FNES)
 Craig Rafter
Craig RafterPostgraduate Research Student, Engineering Sciences (FEE)
 Hossam Ragheb
Hossam RaghebPostgraduate Research Student, Engineering Sciences (FEE)
 Álvaro Ruiz-Serrano
Álvaro Ruiz-SerranoPostgraduate Research Student, Chemistry (FNES)
 Kieran Selvon
Kieran SelvonPostgraduate Research Student, Engineering Sciences (FEE)
 Ashley Setter
Ashley SetterPostgraduate Research Student, Engineering Sciences (FEE)
 Valerio Vitale
Valerio VitalePostgraduate Research Student, Electronics and Computer Science (FPAS)
 Jonathon Waters
Jonathon WatersPostgraduate Research Student, Engineering Sciences (FEE)
 Thorsten Wittemeier
Thorsten WittemeierPostgraduate Research Student, Engineering Sciences (FEE)
 Emanuele Zappia
Emanuele ZappiaPostgraduate Research Student, Engineering Sciences (FEE)
 Petrina Butler
Petrina ButlerAdministrative Staff, Research and Innovation Services
 Susanne Ufermann Fangohr
Susanne Ufermann FangohrAdministrative Staff, Civil Engineering & the Environment (FEE)
 Jan Kamenik
Jan KamenikAlumnus, University of Southampton
Zheng JiangNone, None