Investigation of gas adsorption of metal-organic frameworks using quantum mechanics and Monte Carlo simulation
- Investigators
- Jia Huo
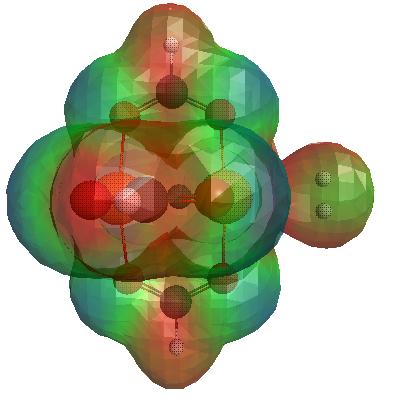
3D Total Electron Density of H2_CuO cluster, optimised by GAMESS and rendered by wxMacMolPlt
Metal organic frameworks (MOFs) has received much attention in the field of gas storage/separation, catalysis, etc, due to their highly ordered porosity, high surface area, multi functionality, chemically talorability and high loading of meta sites. Experimental method has contributed to these areas, but there are still plenty of problems not solved solely from experiment, including investigation of mechanism of adsorption and screening MOFs for target-specific applications. In this project, we plan to use quantum mechanics and Monte Carlo simulation to investigate the various guest adsorption properties on MOFs to screen the substrates catalysed by active sites within MOFs and the influence of transition metal sites of MOFs on gas adsorption for design of MOFs with high gas storage capacity.
Categories
Physical Systems and Engineering simulation: Climate, Energy, Materials
Simulation software: GAMESS(US)