
Michael Carter
- Position
- Postgraduate Research Student
- Institution
- Chemistry (FNES)
- Contact
- Complete this online contact form to contact Michael.
The estimation of ligand binding affinities using computer simulations has now reached a point where it is no longer constrained by underlying free energy methodology, but instead by the reliability of the forcefields and sampling.
To address this problem, the collaboration between the University of Southampton and Novartis UK, will develop a QM/MM based approach to calculate binding free energies, for protein ligand systems, in which the ligand is represented by Density Functional Theory (DFT) and the protein and solvent are represented by point charges within our QM/MM calculations (Fig.1).
Initial work focused on the accurate prediction of hydration free energies for 108 small organic molecules.
Gallery
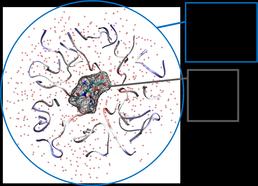
Figure 1: Representation of QM/MM system for Neuraminidase with the inhibitor Oseltamivir (Tamiflu (C)) bound