Large-Scale Quantum Chemistry Simulations of Organic Photovoltaics
- Started
- 5th October 2015
- Research Team
- Gabriele Boschetto
- Investigators
- Chris-Kriton Skylaris
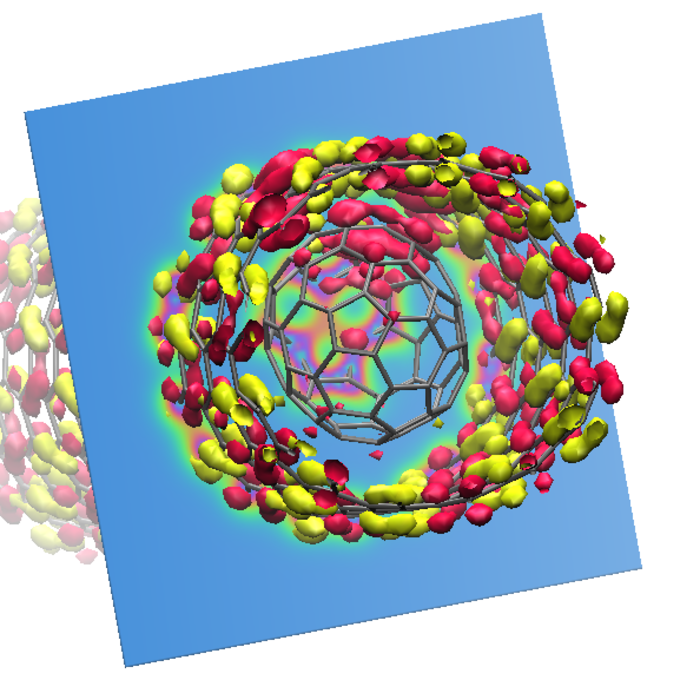
Highest occupied and lowest unoccupied molecular orbitals of a fullerene in a carbon nanotube.
With growing global energy demands and pressure to switch to renewable energy sources, solar photovoltaics (PV) appear particularly attractive, not least because the power received by Earth from the Sun exceeds human power consumption by three orders of magnitude. Organic photovoltaics (OPV) offer certain advantages as compared to established PV technologies such as silicon panels. Organic cells can be lightweight and flexible, thus allowing greater freedom of design and low installation costs. Moreover, while current OPVs are only nearly half as efficient as silicon in full sunlight, they are more effective in diffuse light and indoors, where silicon panels struggle to perform.
A great deal of research effort has been devoted to studying OPV physics, but the connection between molecular structure of materials and device performance is still far from clear. Computer modelling of OPVs has largely been limited to on the one hand to phenomenological or coarse-grained simulations which lose the molecular picture from sight and on the other hand to QM calculations on model small molecules in vacuum. The most widely applied design principles (based on Scharber model) consider energy levels of individual materials separately - either measured for pure materials or calculated for isolated molecules. However, the energy levels of the donor and the acceptor change in different environments, and this difference is the driving force for charge separation (creation of free charge carriers) in high-performance OPVs.
This PhD project will use first principles quantum mechanical calculations to provide a detailed atomic-level understanding of OPV materials and models of bulk heterojunctions on a far larger scale than possible before by using the ONETEP program for linear-scaling first principles quantum mechanical calculations. The studies will utilise several of the capabilities of ONETEP, such as structure optimizations and molecular dynamics simulations of assemblies of oligomer chains with fullerenes, to elucidate the dependence of polymers' IP, EA and Eg on chain length and proximity of fullerene; as well as relationships between molecular structure and packing. The simulations will also involve excited state calculations on polymer chains and polymer-fullerene assemblies, to build a realistic model of light harvesting and charge generation. As state-of-the-art and emerging simulation technologies will be used for this work it is expected that the project will also involve some method development within the ONETEP code.
This project is part of the CDT for Next Generation Computational Modelling (NGCM) and is in collaboration with Merck Chemicals Ltd. Chilworth Technical Centre, Southampton.
Categories
Physical Systems and Engineering simulation: Advanced Materials, Energy, Materials
Algorithms and computational methods: Density functional Theory, Quantum Chemistry
Simulation software: Onetep
Transdisciplinary tags: NGCM