A composite likelihood approach to genome-wide data analyses.
- Started
- 15th September 2008
- Ended
- 14th September 2011
- Research Team
- Jane Gibson, Ioannis Politopoulos
- Investigators
- Andrew Collins
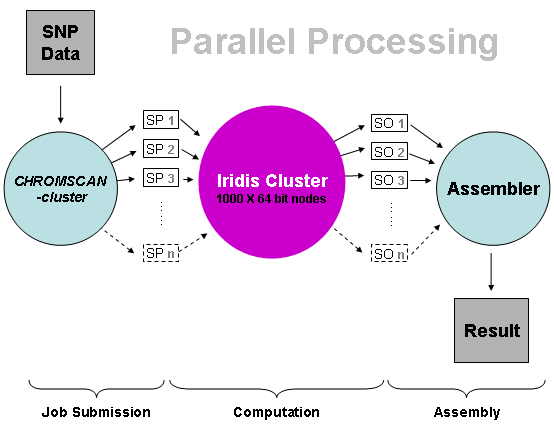
Parallel processing in CHROMSCAN.
We describe composite likelihood-based analysis of a genome-wide breast cancer case-control sample from the Cancer Genetic Markers of Susceptibility project. We determine 14,380 genome regions of fixed size on a linkage disequilibrium map which delimit comparable levels of linkage disequilibrium. Although the numbers of SNPs are highly variable each region contains an average of ~35 SNPs and an average of ~69 after imputation of missing genotypes. Composite likelihood association mapping yields a single P-value for each region, established by a permutation test, along with a maximum likelihood disease location, standard error and information weight. For single SNP analysis the nominal P-value for the most significant SNP (msSNP) requires substantial correction given the number of SNPs in the region. Therefore imputing genotypes may not always be advantageous for the msSNP test, in contrast to composite likelihood. For the region containing FGFR2 (a known breast cancer gene) the largest chi-square is obtained under composite likelihood with imputed genotypes (chi-square increases from 20.6 to 22.7), and compares to a single-SNP based chi-square of 19.9 after correction. Imputation of additional genotypes in this region reduces the size of the 95% confidence interval for location of the disease gene by ~40%. Amongst the highest ranked regions, SNPs in the NTSR1 gene would be worthy of examination in additional samples. Meta-analysis, which combines weighted evidence from composite likelihood in different samples, and refines putative disease locations, is facilitated through defining fixed regions on an underlying linkage disequilibrium map. Technically speaking, genotypic data are split into chromosomal files, i.e. genotypes on one chromosome are analysed each time, requiring on average only 20hrs on Iridis 3. It should be noted that individual completion times vary significantly because of the increased genotypic coverage especially in larger chromosomes. Nevertheless, Iridis allows for parallel analyses of all chromosomes and, therefore, reduces the overall completion time to the time required for the largest chromosome. To further validate findings, we are also currently analysing genotypic data from other cohorts such as the Welcome Trust Case Conrol Consortium (WTCCC) and the Prospective study of Outcomes in Sporadic versus Hereditary (POSH) breast cancer. Additionally, we are currently setting up pipelines to analyse exome and whole-genome data as, in the next few months, we will be receiving several dozens of exomes, each of which consists of approximately 30 gigabases of DNA sequence at 30x "depth" or, in other words, 30*30=900 gigabases. In the case of whole-genome data numbers are significantly higher, at least 100 times as much as in that of exome data. Such voluminous data require not only enormous storage capacity but also powerful computational resources and this is a great opportunity to make the most out of Iridis 3 in that respect.
Categories
Life sciences simulation: Bioinformatics, Epidemiology, NextGen Sequencing
Programming languages and libraries: C++
Computational platforms: Iridis