Water Molecules in Protein Binding Sites
- Research Team
- Michael Bodnarchuk
- Investigators
- Jonathan Essex
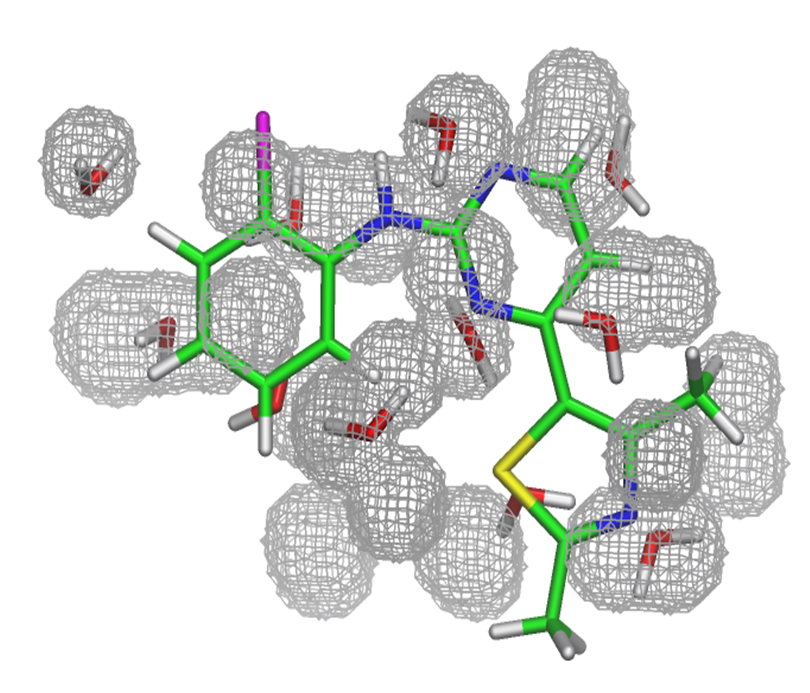
Predicted water molecules for the CDK2 apo binding pocket, found using GCMC. The crystallographic ligand is shown for clarity
The importance of water molecules in drug design and protein structures has become of considerable interest in the last 10-15 years. Traditionally, it has been thought that there are two major roles that water molecules play in ligand binding. The first role is to stabilise a protein-ligand complex through creating a hydrogen bonding network, seen in systems such as N9-neuraminidase. The second role is the ability of a water molecule to be displaced upon ligand binding, seen in systems such as HIV-1 protease. This is typically advantageous since the release of an ordered water molecule into the bulk carries an entropic gain, coupled with an enthalpic gain of strong protein-ligand interactions.
More recent studies have helped to shed even more light upon the multiple, and often complex, roles in which water molecules play in protein binding sites. Seemingly subtle changes in water-based hydrogen bonding networks have been shown to affect ligand-protein interaction energies, highlighting the need for an accurate representation of solvation within a protein binding site. Studies by Setny, Baron and McCammon have shown that water molecules play an active role in determining ligand-protein binding or rejection, whether this be due to mediating direct interactions, or providing an electrostatic screening effect. A recent communication by Shan has also shown that water molecules can form a solvation shell between the protein and ligand in the binding site prior to the binding event, with the formation of this solvation shell providing a kinetic barrier to binding.
As such, it is evident that a realistic hydration pattern is vital if protein-ligand interactions are to be fully understood. Crystallographic approaches are often limited in their ability to finding water molecules due to resolution issues and crystallographer preference, requiring an alternative method to be employed. Our work is focussed upon developing methods and procedures aimed towards creating realistic hydration patterns, typically using the Just Add Water Molecules (JAWS) and Grand Canonical Monte Carlo (GCMC) theories. These methods allow us to both predict, and also score, the location of water molecules in a variety of systems.
Categories
Life sciences simulation: Bioinformatics, Biomolecular simulations
Algorithms and computational methods: Molecular Mechanics, Monte Carlo
Simulation software: AMBER, ProtoMS
Visualisation and data handling software: VMD
Programming languages and libraries: Fortran
Computational platforms: Iridis