Development and application of powerful methods for identifying selective sweeps
- Started
- 2nd October 2017
- Research Team
- Clare Horscroft
- Investigators
- Andrew Collins, Reuben Pengelly, Timothy Sluckin, Sarah Ennis
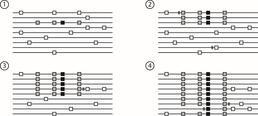
A selective sweep in progress over four generations. The black square in (1) is a new beneficial mutation. (2) and (3) show the mutation spreading through the population, with the two grey alleles hitchhiking along with it. (4) shows the final population.
Around 10,000 years ago, humans started to domesticate cows. It was also around this time that humans started to drink cow’s milk, which provided an advantage to those people who had the genetic mutation giving them the ability to process lactose into adulthood. Evidence of this selective advantage can be found in the DNA of some populations of humans today. Other examples of selected traits found in some human populations include disease resistance and adaptation to live at high-altitudes.
Genetic signatures are left in the DNA of a population when natural selection or evolution occurs. As a selected trait sweeps through a population (generation by generation), DNA strings around the site of selection “hitchhike” along. This creates areas of the genome which are highly correlated: that is, if you have one allele, you are very likely to have another particular allele nearby. These signatures can be identified using mathematical models designed to locate these areas of unusually high correlation.
This PhD project involves the development and application of models to population genomic data to find evidence of selective sweeps in humans and also in other species, such as chickens. This will involve interrogating huge datasets of whole genome sequences for large populations (for example, the 100,000 Genomes Project), requiring the use of the Iridis cluster to manage and process these large datasets. In addition, developing and testing methods will necessitate large simulation runs and analysis which will also require high performance computing capabilities.
Categories
Life sciences simulation: Biomathematics, Evolution
Algorithms and computational methods: Machine learning
Software Engineering Tools: RStudio
Programming languages and libraries: R
Computational platforms: Iridis
Transdisciplinary tags: IfLS