Dipole moment and theoretical spectroscopy: a computational approach
- Started
- 1st March 2013
- Ended
- 16th May 2013
- Research Team
- Valerio Vitale
- Investigators
- Chris-Kriton Skylaris
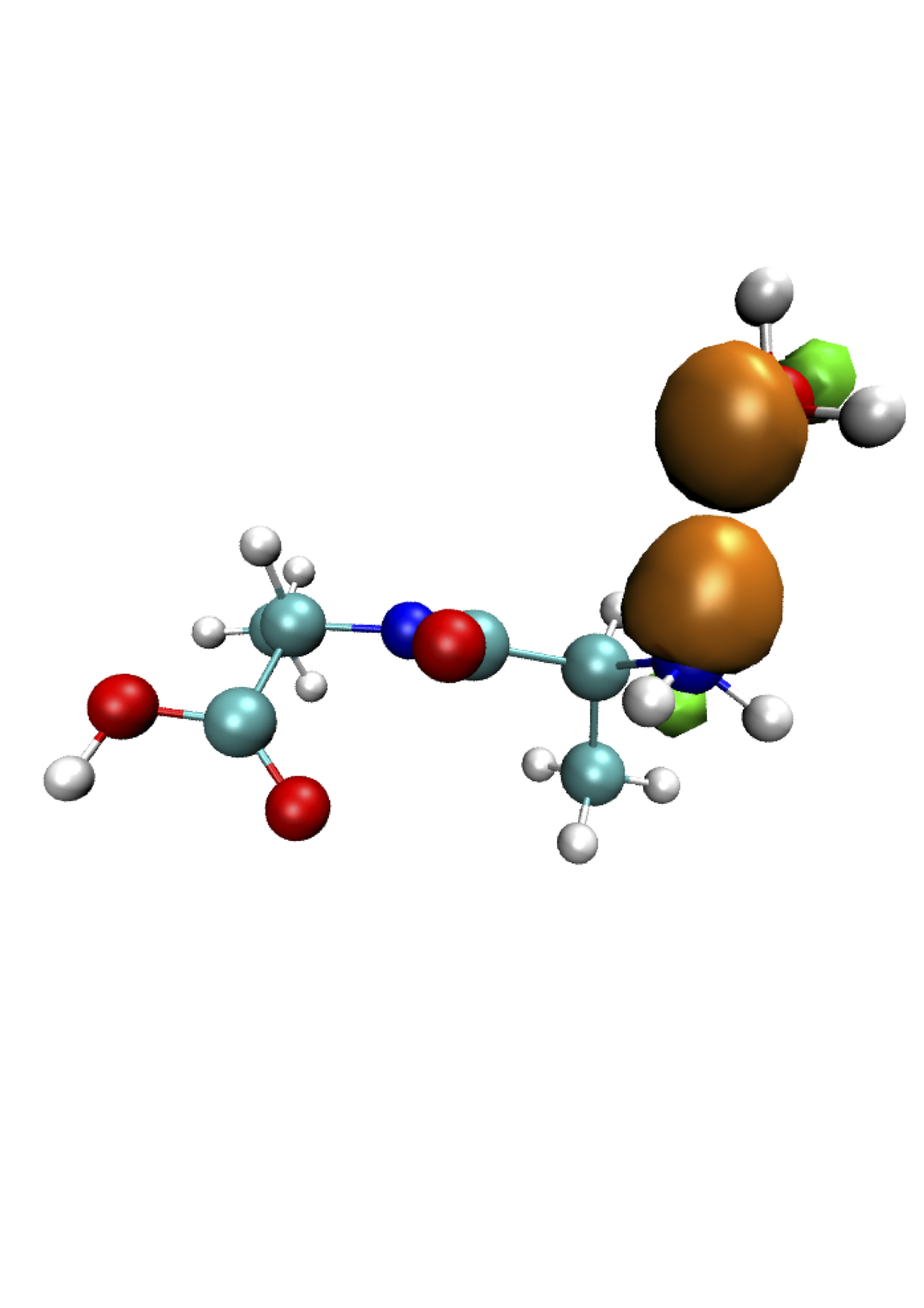
Few Non-orthogonal Generalised Wannier Functions for a charged dialanine molecule (Ala2H+) and a water molecule (H2O) in gas phase
The present project represents a first step towards the implementation of a new technique to calculate the whole vibrational spectra of molecules in a formally exact way, which fully takes into account anharmonicity and conformational transitions, at a finite temperature, both in gas phase and in solution in a single molecular dynamics simulation, combining the features of ONETEP[3] (linear-scaling DFT) and a method introduced by M. P. Gaigeot and her group [4], [5]. This technique is based on the calculation of the Fourier transform of the dipole moment autocorrelation function (FTTCF). Although the FTTCF formalism is a standard procedure for the calculation of several properties in classical molecular dynamics, it usually is not very accurate in predicting molecular properties due to the classical framework. Now we are able to use the FTTCF formalism in a more accurate way, by treating the electronic interactions directly through the foundamental equations of quantum mechanics. In order to evaluate the vibrational spectra we must be able to compute the transition dipole moment of our systems. This can be a tricky point for quantum systems when adopting periodic boundary conditions (PBC), as we do in ONETEP. Therefore, for this project we set the field for future calculations, by implementing in ONETEP a code to calculate the total dipole moment of molecules. The results have been used in a subsequent project, where the vibrational spectra of small molecules have been computed.
[1] M. Richard Martin. Electronic Structure. Basic Theory and Practical Methods. Cambridge University Press, 5th printing edition, 2011.
[2] Physics Today, 58(6):49–54, 2005.
[3] C.-K. Skylaris, P. D. Haynes, A. A. Mostofi and M. C. Payne, J. Chem. Phys. 122, 084119 (2005)
[4] Phys. Chem. Chem. Phys., 12:3336–3359, 2010
[5] The Journal of Physical Chemistry A, 110(28):8802–8810, 2006.
Categories
Physical Systems and Engineering simulation: Materials, Quantum Dynamics
Algorithms and computational methods: Density functional Theory, Quantum Chemistry
Simulation software: Onetep
Visualisation and data handling software: VMD
Programming languages and libraries: Fortran, MPI, OpenMP
Transdisciplinary tags: Complex Systems