Mass Spec identification of proteins utilising EST libraries
- Homepage
- http://www.soton.ac.uk/~re1u06/software/budapest/index.html
- Started
- 24th January 2008
- Research Team
- Bethan Jones
- Investigators
- Richard Edwards, Maria Debora Iglesias-Rodriguez
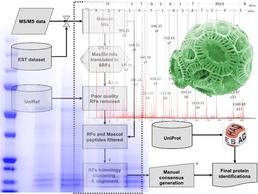
Mass spec identification using EST data opens up proteomics investigations to organisms without sequenced genomes.
Despite advances in sequencing technology, we are still a long way from having well-annotated genome sequences for all organisms of interest. Instead, EST (Expressed Sequence Tag) data is available for less popular organisms in many cases, prior to a genome project. EST data presents a particular challenge for the identification of proteins using mass spectrometry (MS): it is often redundant (multiple copies of the same gene), consists primarily of short fragments of coding sequence, contains many sequencing errors and is generally poorly annotated. Nevertheless, generating EST libraries represents a much cheaper alternative to full genome sequence and has the advantage of focusing on the parts of the genome of interest to proteomics, i.e. the expressed genes, many of which will be protein-coding.
The School of Biological Sciences, Centre for Proteomic Research and the National Oceanography Centre, Southampton have been collaborating to develop bioinformatics analysis tools to help clean up the results of searching MS against EST databases to identify proteins. This has resulted in the development of BUDAPEST (Bioinformatics Utility for Data Analysis of Proteomics on ESTs), which translates, assembles and annotates ESTs identified during a MASCOT search of MS data to generate a high quality non-redundant results dataset with the required peptide support.
Categories
Life sciences simulation: Bioinformatics, Evolution
Programming languages and libraries: Python