The hydrogen abstraction phase of the CYP-cyclohexene reaction, using large-scale DFT
- Homepage
- http://www.onetep.org/
- Started
- 9th January 2012
- Research Team
- Chris Pittock, Karl Wilkinson
- Investigators
- Chris-Kriton Skylaris
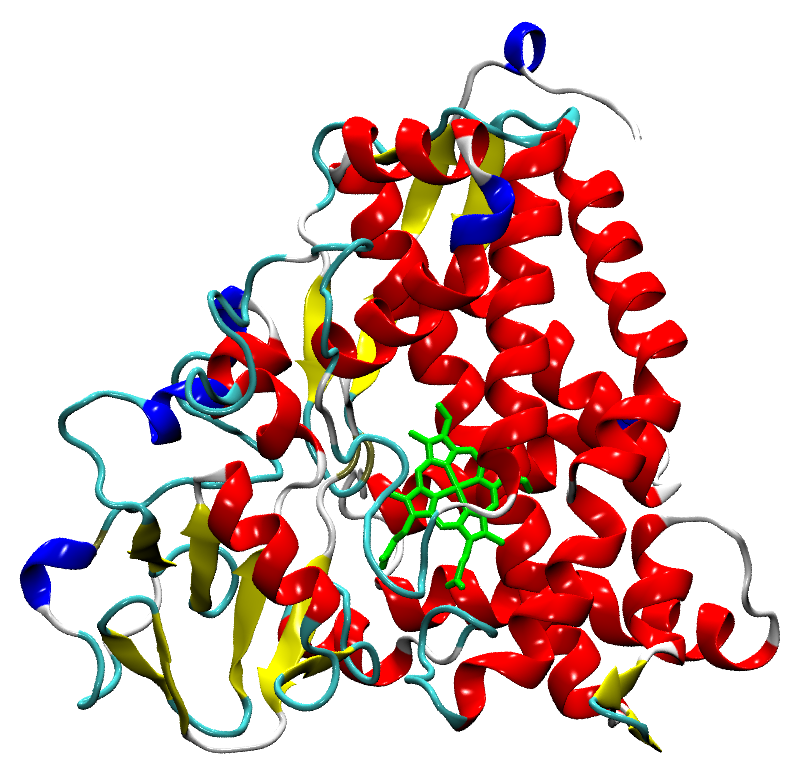
Tertiary structure of cytochrome P450_cam (PDB code: 1DZ9), shown in the 'Cartoon' method. The haem molecule that is part of Compound I is highlighted in green.
Cytochrome P450 (CYP) is present in almost all forms of life, and is responsible for the oxidation of small molecules, as part of the metabolism process. In the human body, CYP in the liver oxidises drug molecules into a more soluble form that makes it easier to excrete. In the majority of cases, this product is non-toxic and a trivial matter, but there are some cases where CYP-based oxidation produces a toxic byproduct. If manual or biological intervention cannot resolve this, protein damage can occur (notably liver damage). With this in mind, the pharmaceutical industry would benefit greatly from a method of predicting the metabolism products of any molecule, as part of their drug discovery process.
Predictive models already exist, but despite the promising results[1] either rely on a database of experimentally-derived results, or are severely limited to only one of the twelve different types of CYP in the human body - a dynamic method that would represent any substrate with any CYP isoform could be possible if the high-accuracy quantum mechanics (QM) method of energy calculation is used. The challenge comes in accurately modelling the active site: a six-coordinate Fe(IV), triradical haem complex (Compound I), and through the wide range of previous research into the electronic properties of Compound I[2], we hope to calculate the transition state of the hydrogen-abstraction reaction between cyclohexene and Compound I. However, instead of the 'norm', where only 50-100 atoms are modelled with QM (one example being in [3]), we increase the scale to 500 and 1000 atoms to observe the role that the protein has in manipulating the metabolism activity of CYP.
Modelling a system of such a large size is not possible through conventional QM methods, and requires a linear-scaling method such as ONETEP (part-developed by the Skylaris Group, Southampton) to achieve. Regardless of this, a significant amount of computational resources are required to 'up the ante' by adding proteinous atoms. This is where the use of such large HPC resources, such as Iridis is essential towards our work.
[1] - Stjernschantz, E.; Vermeulen, N.P.E.; Oostenbrink, C. Expert Opin. Drug Metab. Toxicol. 2008, 4(5), 513-527.
[2] - Shaik, S.; Cohen, S.; Wang, Y.; Chen, H.; Kumar, D.; Thiel, W. Chem. Rev. 2010, 110, 949-1017.
[3] - Lonsdale, R.; Harvey, J.N.; Mulholland, A.J.; J. Phys. Chem. Lett. 2010, 1(21), 3232-3237.
Categories
Life sciences simulation: Biomolecular simulations
Algorithms and computational methods: Density functional Theory, Optimisation, Quantum Chemistry