Water molecules in drug development: can we predict drug affinity when water molecules are involved?
- Homepage
- http://www.essexgroup.soton.ac.uk/ProtoMS/
- Started
- 1st October 2015
- Research Team
- Hannah Bruce Macdonald, Christopher Cave-Ayland
- Investigators
- Jonathan Essex
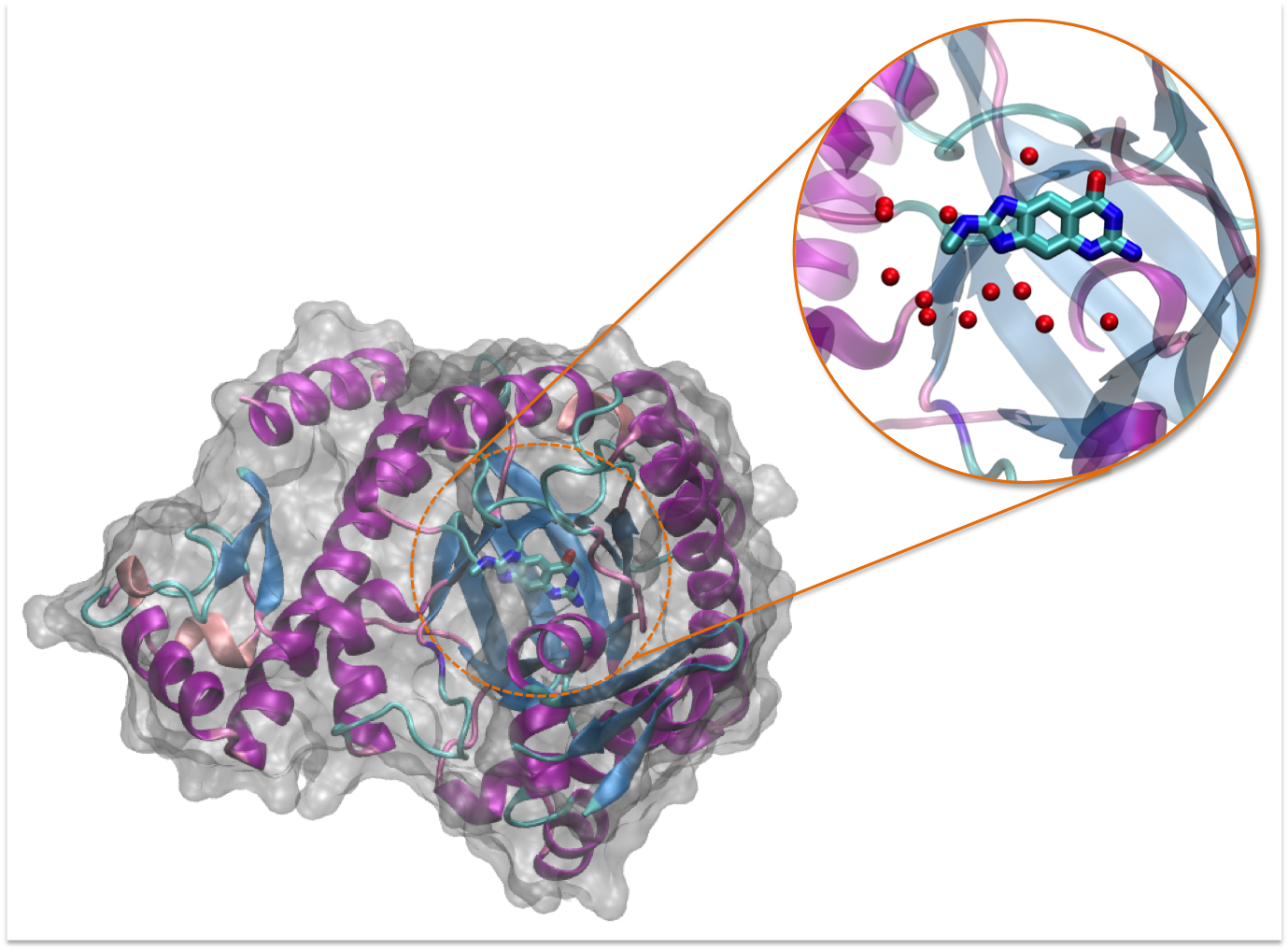
Protein with drug molecule bound. Red spheres show locations of water molecules which can help the drug interact favourably with the protein.
Water is the biological solvent and is involved in a huge range of biological processes. One of these processes is drug action; when a drug binds to a protein, water molecules can remain in the protein and form stable interactions with the drug, increasing its effectiveness. We aim to predict two factors; the location of water molecules in these systems, and the relative energy of different drug molecules to a protein.
Locating water molecules and calculating drug affinities can be difficult experimentally, and computational simulations can help. We are able to simulate the complex protein systems using our in-house code ProtoMS on the Iridis supercomputer. To calculate the location of water molecules, the simulations require many ‘repeats’ running simultaneously, each with very small differences in the system. The OpenMPI libraries available on the supercomputer are able to facilitate this.
Simulations to calculate the binding energy of drugs are computationally demanding. We have developed a method that is able to calculate the water locations and the drug affinity simultaneously. This requires generating a 2-dimensional network of simulations, which requires multiple CPU nodes to perform. One ‘dimension’ of the simulation is able to monitor the effect of the water molecules, and the other observes the different energies of the drugs.
Categories
Life sciences simulation: Biomedical, Biomolecular simulations
Simulation software: AMBER, ProtoMS
Visualisation and data handling software: VMD
Software Engineering Tools: Mercurial
Programming languages and libraries: Fortran, MPI, Python
Computational platforms: Iridis, Linux
Transdisciplinary tags: Scientific Computing