Seminar 26th April 2013 4 p.m. 85/2009
Modelling hydration in simple and complex systems with inhomogeneous fluid solvation theory
Dr David Huggins
University of Cambridge
- Categories
- AMBER, Bioinformatics, Biomathematics, Biomedical, Biomolecular Organisation, Biomolecular simulations, C, CASTEP, Complex Systems, Computer Science, CUDA Fortran, Developmental Biology, Economics, Environmental hazards, Fortran, Gaussian, GPU-libs, Gromacs, Heat transfer, HECToR, HPCx, Iridis, Jaguar, Linux, Liquid crystals, Lyceum, Medical Imaging, MEMS, Monte Carlo, Multi-core, Multi-physics, Multi-scale, Multipole methods, Nanoscale Assemblies, NWCHEM, OpenCL, OpenMP, Optimisation, PETSc, Quantum Chemistry, Scientific Computing, Software Engineering, Structural biology, Systems biology, Visualisation
- Submitter
- Chris-Kriton Skylaris
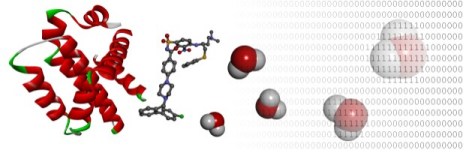
Water simulations
Water molecules are a key component of biological systems and act as ordered structural elements at binding interfaces. The mediation of ligand binding by water molecules can have important consequences for binding affinity and specificity. The binding of small molecules to proteins is accompanied by displacement of water molecules from protein hydration sites. The thermodynamic properties of these hydration sites can be predicted using the statistical mechanical method of inhomogeneous fluid solvation theory (IFST). We have analysed the predictions of IFST based on molecular dynamics simulations of Hsp90 and compared them with the crystallographic binding modes of small molecules. This has allowed us to assess druggability and provide retrospective explanations for experimental SAR and observed binding modes. In further work, we have assessed the quantitative accuracy of IFST using solvation free energies of small molecules.