Measuring biomolecules - improvements to the spectroscopic ruler
- Started
- 1st June 2009
- Research Team
- Jan Junis Rindermann, James Richardson
- Investigators
- Pavlos Lagoudakis, Tom Brown
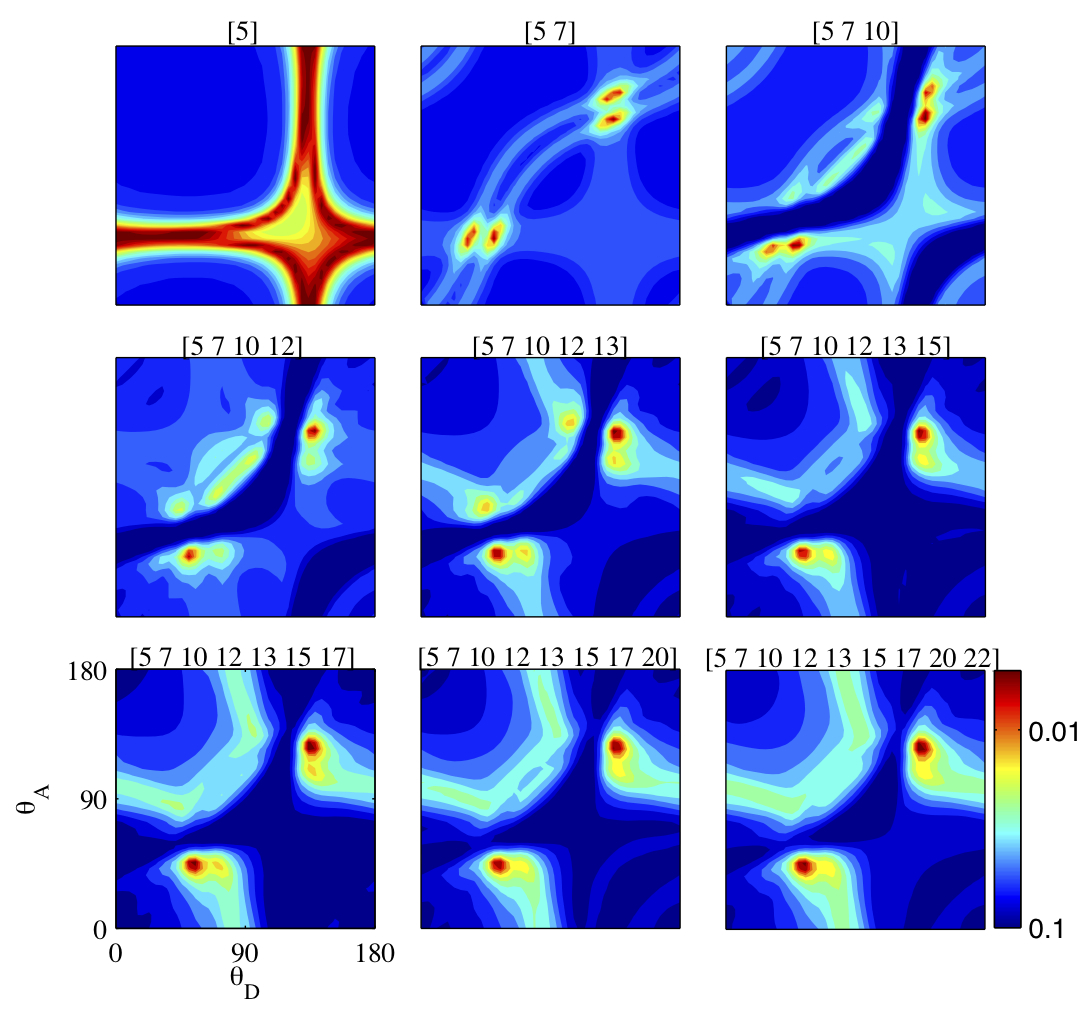
Scans of chi-square through the parameter space in the vicinity of the best fit parameters. From the top left to the bottom right image the constraints in the fitting routine are increased and the fit converges to a unique point (except for a symmetry).
The spectroscopic ruler is a technique to measure the geometry of biomolecules on the nm scale by labeling them with pairs of fluorescent markers and measuring distance dependent non-radiative energy transfer between them. The technique can be applied in the living cell and is widely used for bio-medical research. The remaining uncertainty and most common source of errors originates from the unknown orientation between the optical dipole moments of the fluorescent markers, especially when the molecule undergoes thermal fluctuations in physiological conditions. Recently we introduced a simulation based method for the interpretation of the fluorescence decay dynamics of the markers that allows us to retrieve both the average orientation and the extent of directional fluctuations of the involved dipole moments (Rindermann et al., J. Am. Chem. Soc., Article ASAP (2010)).
In this project we study artificially synthesised DNA helices which are labelled with donor and acceptor fluorophores in nine different configurations. The DNA helices serve as nano-scaffolds to keep the optical dipole moments of the fluorophores in a controlled geometry, a requirement to test our methodology. Non-radiative energy transfer (also known as Fluorescence Resonance Energy Transfer (FRET)) is studied by means of ultrafast spectroscopy to obtain the fluorescence decay curves of the quenched donor species. For the simulation of FRET, which is mediated via dipole-dipole coupling, the directional fluctuations of the dipole moments are accounted for by considering a von Mises-Fisher distribution for their orientation and a set of geometrical parameters of the DNA molecule. A global fitting routine analyses simultaneously the experimental data from all samples and yields the relevant characteristic geometrical parameters of the fluorescent markers. This methodology allows an accurate read out of the distance between the fluorescent markers from the fluorescence decay curves.
During data analysis a large numbers of simulations has to be carried out because a priori it is not possible to exclude or favour particular configurations of the molecule. Iridis is used to effectively run multiple fitting routines in parallel, starting from different points in the nine dimensional parameter space: a task too big for a desktop PC. After the random exploration of the parameter space the local vicinity of the best fit parameters is tested by changing the constraints on the fitting routine. The results of these scans demonstrate that albeit the apparently high degree of freedom our method provides meaningful output in the form of unique parameters describing FRET in the molecules under investigation.
After the demonstration of the feasibility of our approach we are currently working on a public interface to the simulation. It will allow the application of the technique to a wide range of different molecular geometries and configurations.
Team members: This project is a collaboration between Jan Junis Rindermann and Prof. Pavlos Lagoudakis from the School of Physics, Dr. James Richardson and Prof. Tom Brown from the School of Chemistry and Dr. Yosef Akhtman from the School of Electronics and Computer Science at the University of Southampton.
Categories
Life sciences simulation: Biomedical, Biomolecular simulations, Nanoscale Assemblies
Physical Systems and Engineering simulation: Photonics
Algorithms and computational methods: Optimisation
Software Engineering Tools: SVN
Programming languages and libraries: C++, Matlab
Computational platforms: Iridis, Linux, Mac OS X, Windows
Transdisciplinary tags: Scientific Computing