Molecular Fragments in Inhibitor Design
- Started
- 15th April 2010
- Research Team
- Michael Bodnarchuk
- Investigators
- Jonathan Essex
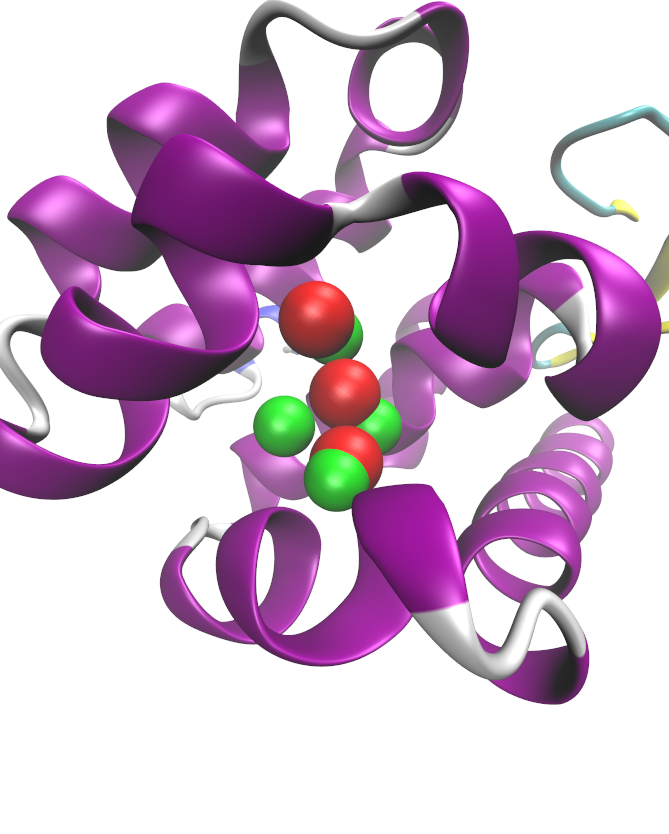
JAWS prediction for the location of water molecules in the hydrophobic T4-lysozyme L99A binding pocket. Crystallographic waters are shown in red, with the JAWS prediction in green
Fragment-Based Drug Discovery (FBDD) has shown great promise as a tool in the medicinal chemists' toolbox. Instead of searching the entire realm of chemical space the method relies on the fact that drug molecules are generally made up of a finite number of building blocks, known as fragments. As a result, searching for these fragments becomes a much more tractable task than randomly trialling existing drug libraries. The results obtained from FBDD can be used to create drug molecules which are both more potent and selective, utilising the fragments as building blocks.
Whilst performing FBDD experimentally yields direct results, the experiments are often costly and time consuming. In order to overcome this barrier, computational methods can be used to try and simulate the fragment binding process. Existing FBDD methods typically have two major drawbacks. Firstly the protein and fragments are typically modelled as rigid, whilst it is well established that subtle conformational changes in the protein side chains can directly influence binding. Secondly the competition with solvent is typically ignored, resulting in results which do not account for the fact that the fragment might not exist in the protein binding site and would lie in the aqueous bulk.
Our work is hence focussed on trying to overcome these barriers and forming a method which can predict fragment binding a priori, whilst taking into account solvation and flexibility effects. The method being utilised in this project, based upon the Just Add Water Molecules (JAWS) method, has use in not only FBDD but also water molecule placement and binding free energy prediction. To this end we have performed comparative simulations between JAWS and existing rigorous free energy calculations, and shown that JAWS gives excellent agreement whilst considerably reducing the computational cost.
Categories
Life sciences simulation: Biomolecular simulations
Algorithms and computational methods: Molecular Dynamics, Monte Carlo
Simulation software: AMBER, ProtoMS
Visualisation and data handling software: Xmgrace
Programming languages and libraries: Fortran