Molecular Dynamics
The molecular dynamics method.
For queries about this topic, contact Syma Khalid.
View the calendar of events relating to this topic.
Projects

Ab initio simulations of chemical reactions on platinum nanoparticles
Chris-Kriton Skylaris (Investigator), Álvaro Ruiz-Serrano, Peter Cherry
•Use first principles calculations to study the relationship between shape and size of nanoparticle and the oxygen adsorption energy.
• Investigate the effect of high oxygen coverage on the catalytic activity of the nanoparticles.
Can we calculate the pKa of new drugs, based on their structure alone?
Chris-Kriton Skylaris (Investigator), Chris Pittock, Jacek Dziedzic
The pKa of an active compound in a pharmaceutical drug affects how it is absorbed and distributed around the human body. While there are various computational methods to predict pKa using only molecular structure data, these tend to be specialised to only one class of drug - we aim to generate a more generalised prediction method using quantum mechanics.

Centre for Doctoral Training in Next Generation Computational Modelling
Hans Fangohr, Ian Hawke, Peter Horak (Investigators), Susanne Ufermann Fangohr, Thorsten Wittemeier, Kieran Selvon, Alvaro Perez-Diaz, David Lusher, Ashley Setter, Emanuele Zappia, Hossam Ragheb, Ryan Pepper, Stephen Gow, Jan Kamenik, Paul Chambers, Robert Entwistle, Rory Brown, Joshua Greenhalgh, James Harrison, Jonathon Waters, Ioannis Begleris, Craig Rafter
The £10million Centre for Doctoral Training was launched in November 2013 and is jointly funded by EPSRC, the University of Southampton, and its partners.
The NGCM brings together world-class simulation modelling research activities from across the University of Southampton and hosts a 4-year doctoral training programme that is the first of its kind in the UK.
Computational chemistry study on the interaction mechanism of imidazolium based ionic liquid lubricants with metal surface
Ugur Mart (Investigator)
We propose a fundamental research to investigate the interaction mechanism of ionic liquids (ILs) with metal surfaces, molecular structure and organization on the surface along with chemical reactions using computational chemistry methods at molecular level.
Development of wide-ranging functionality in ONETEP
Chris-Kriton Skylaris (Investigator), Jacek Dziedzic
ONETEP is at the cutting edge of developments in first principles calculations. However, while the fundamental difficulties of performing accurate first-principles calculations with linear-scaling cost have been solved, only a small core of functionality is currently available in ONETEP which prevents its wide application. In this collaborative project between three Universities, the original developers of ONETEP will lead an ambitious workplan whereby the functionality of the code will be rapidly and significantly enriched.

Dual resolution simulations of lipid membrane systems
Jonathan Essex (Investigator), Kieran Selvon
This project aims to shed light on cell membrane mechanisms which are difficult to probe experimentally, in particular drug permiation across the cell membrane. If one had a full understanding of this mechanism, drugs could be designed to easily cross the membrane, or target particular embedded proteins to improve their efficacy. A reliable and robust computational method to asses a molecules permeability would be invaluable in the field of drug design, we seek to perfect such a method.
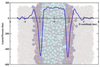
How far can we stretch the MARTINI?
Syma Khalid (Investigator), Ric Gillams
To date, coarse-grained lipid models have generally been parameterised to ensure the correct prediction of structural properties of membranes, such as the area per lipid and the bilayer thickness. The work described here explores the extent to which coarse-grained models are able to predict correctly bulk properties of lipids (phase behaviour) as well as the mechanical properties, such as lateral pressure profiles and stored elastic stress in bilayers. Such an evaluation is crucial for understanding the predictive capabilities of coarse-grained models.
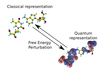
Hybrid quantum and classical free energy methods in computational drug optimisation
Jonathan Essex, Chris-Kriton Skylaris (Investigators), Christopher Cave-Ayland
This work is based around the application of thermodynamics and quantum mechanics to the field of computational drug design and optimisation. Through the application of these theories the calculation of the physical properties of drug-like molecules is possible and hence some predictive power for their pharmaceutical activity in vivo can be obtained.
Immunotherapy Research: Modelling MHC Class I Complex Assembly
Timothy Elliott, Jorn Werner (Investigators), Alistair Bailey
This project uses mathematical modelling and simulation to investigate mechanisms by which our cells process and present biological information that is used by our immune system to distinguish between healthy and diseased cells.

Investigation into the Interfacial Physics of Field Effect Biosensors
Nicolas Green, Chris-Kriton Skylaris (Investigators), Benjamin Lowe
This interdisciplinary research aims to improve understanding of Field Effect Transistor Biosensors (Bio-FETs) and to work towards a multiscale model which can be used to better understand and predict device response.
Lyotropic phase transitions of lipids studied by CG MD simulation and experimental techniques
Syma Khalid (Investigator), Josephine Corsi
A study of the phase behaviour of cationic lipid - DNA complexes such as those used for transfection by coarse grained molecular dynamics simulation. Lipid systems studied include DOPE, DOPE/DNA and DOPE/DOTAP/DNA. Structural parameters and phase behaviour observed computationally have been compared with those gained using Small Angle X-ray Scattering (SAXS) and polarising light microscopy techniques.

Modelling micromagnetism at elevated temperature
Hans Fangohr, Kees de Groot, Peter de_Groot (Investigators), Dmitri Chernyshenko
We aim to develop a multiscale multiphysics model of
micromagnetism at elevated temperatures with atomistic simulations for
material parameter. The tool will be used to guide the development of the next generation magnetic data storage technology: heat assisted magnetic recording.
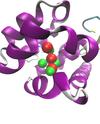
Molecular Fragments in Inhibitor Design
Jonathan Essex (Investigator), Michael Bodnarchuk
Fragment-Based Drug Discovery (FBDD) has emerged as an important tool in the drug discovery process. Instead of screening entire drug molecules, FBDD screens molecular fragments; constituents which make up drug molecules. A computational approach to identifying fragment binding is currently being sought which also yield binding free energy estimation.

Multi-scale simulations of bacterial outer-membrane proteins
Syma Khalid (Investigator), Jamie Parkin
Using Iridis to run multiple simulations, I aim to simulate the outer membrane proteins of Pseudomonas aeruginosa, using X-ray crystal structures of proteins only recently resolved by Bert van den Berg, University of Massachusetts. By modelling the proteins in a realistic P. aeruginosa outer membrane, I aim to gain insight into the binding of these proteins to specific substrates and their function.
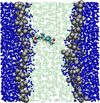
Multiscale modelling of biological membranes
Jonathan Essex (Investigator), Mario Orsi
Biological membranes are complex and fascinating systems, characterised by proteins floating in a sea of lipids. Biomembranes, besides being the fundamental structures employed by nature to encapsulate cells, play crucial roles in many phenomena indispensable for life, such as growth, energy storage, and in general information transduction via neural activity. In this project, we develop and apply multiscale computational models to simulate biological membranes and obtain molecular-level insights into fundamental structures and phenomena.

On the applicability of nonlinear timeseries methods for partial discharge analysis
Paul Lewin (Investigator), Lyuboslav Petrov
The governing processes of Partial Discharge (PD)
phenomena trigger aperiodic chains of events resulting in ’ap-
parently’ stochastic data, for which the widely adopted analysis
methodology is of statistical nature. However, it can be shown,
that nonlinear analysis methods can prove more adequate in
detecting certain trends and patterns in complex PD timeseries.
In this work, the application of nonlinear invariants and phase
space methods for PD analysis are discussed and potential pitfalls
are identified. Unsupervised statistical inference techniques based
on the use of surrogate data sets are proposed and employed for
the purpose of testing the applicability of nonlinear algorithms
and methods. The Generalized Hurst Exponent and Lempel Ziv
Complexity are used for finding the location of the system under
test on the spectrum between determinism and stochasticity. The
algorithms are found to have strong classification abilities at
discerning between surrogates and original point series, giving
motivation for further investigations.

Origins of Evolvability
Richard Watson, Markus Brede (Investigators), William Hurndall
This project examined the putative evolvability of a Lipid World model of fissioning micelles. It was demonstrated that the model lacked evlovability due to poor heritability. Explicit structure for micelles was introduced along with a spatially localised form of catalysis which increased the strength of selection as coupling between potential chemical units of heredity were reduced.

Probing the oligomeric state and interaction surface of Fukutin Transmembrane Domain in lipid bilayer via Molecular Dynamics simulations
Syma Khalid, Philip Williamson (Investigators), Daniel Holdbrook, Jamie Parkin, Nils Berglund, Yuk Leung
Fukutin Transmembrane Domain (FK1TMD) is localised to the endoplasmic reticulum or Golgi Apparatus within the cell where it is believed to function as a glycosyltransferase. Its localisation within the cell is thought to be mediated by the interaction of its N-terminal transmembrane domain with the lipid bilayers surrounding these compartments, each of which possess a distinctive lipid composition. Studies have revealed that the N-terminal transmembrane domain of FK1TMD exists as dimer within dilauroylphosphatidylcholine bilayers and this interaction is driven by interactions between a characteristic TXXSS motif. Furthermore residues close to the N-terminus that have previously been shown to play a key role in the clustering of lipids are shown to play a key role in anchoring the protein in the membrane.
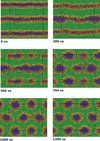
Simulation of biological systems at long length and distance scales
Jonathan Essex (Investigator), Kieran Selvon
This project aims to shed light on cell membrane mechanisms which are difficult to probe experimentally, in particular drug permiation across the cell membrane. If one had a full understanding of the mechanism, drugs could be designed to target particular embedded proteins to improve their efficacy, the viability of nano based medicines and materials could also be assessed, testing for toxicity etc.
Sustainable domain-specific software generation tools for extremely parallel particle-based simulations
Chris-Kriton Skylaris (Investigator)
A range of particle based methods (PBM) are currently used to simulate materials in chemistry, engineering, physics and biophysics. The 4 types of PBM considered directly in the proposed are molecular dynamics (MD), the ONETEP quantum mechanics-based program, discrete element modelling (DEM), and smoothed particle hydrodynamics (SPH).
The overall research objective is to develop a sustainable tool that will deliver, in the future, cutting edge research applicable to applications ranging from dam engineering to atomistic drug design.

The application and critical assessment of protein-ligand binding affinities
Jonathan Essex (Investigator), Ioannis Haldoupis
A method that can accurately predict the binding affinity of small molecules to a protein target would be imperative to pharmaceutical development due to the time and resources that could be saved. A head-to-head comparison of such methodology, ranging from approximate methods to more rigorous methods, is performed in order to assess their accuracy and utility across a range of targets.

THE NORM MATE TRANSPORTER FROM N. GONORRHEAE: INSIGHTS INTO DRUG & ION BINDING FROM ATOMISTIC MOLECULAR DYNAMICS SIMULATIONS
Syma Khalid (Investigator), Daniel Holdbrook, Thomas Piggot, Yuk Leung
The multidrug and toxic compound extrusion (MATE) transporters extrude a wide variety of substrates out of both mammalian and bacterial cells via the electrochemical gradient of protons and cations across the membrane. Multiple atomistic simulation are performed on a MATE transporter, NorM from Neisseria gonorrheae (NorM_NG) and NorM from Vibrio cholera (NorM_VC). These simulations have allowed us to identify the nature of the drug-protein/ion-protein interactions, and secondly determine how these interactions contribute to the conformational rearrangements of the protein.
The ONETEP project
Chris-Kriton Skylaris (Investigator), Stephen Fox, Chris Pittock, Álvaro Ruiz-Serrano, Jacek Dziedzic
Program for large-scale quantum mechanical simulations of matter from first principles quantum mechanics. Based on theory and algorithms we have developed for linear-scaling density functional theory calculations on parallel computers.

Understanding Stochastic Processes in Interacting Spin Models
Oliver Laslett
Applying efficient computational models to compute Langevin dynamics and master equation equilibrium solutions for interacting magnetic spin systems.

Using Molecular Dynamics to Understand the Antibacterial Mechanisms of Daptomycin & Chlorhexidine to Target the Bacterial Membrane
This project aims to use molecular dynamics techniques to understand how antimicrobial peptides, daptomycin and chlorhexidine, disrupt both gram positive and negative cell membranes on an atomic level.

Using Molecular Dynamics to Understand the Antibacterial Mechanisms of Daptomycin & Chlorhexidine to Target the Bacterial Membrane
This project aims to use molecular dynamics techniques to understand how antimicrobial peptides, daptomycin and chlorhexidine, disrupt both gram positive and negative cell membranes on an atomic level.

Using Molecular Dynamics to Understand the Antibacterial Mechanisms of Daptomycin & Chlorhexidine to Target the Bacterial Membrane
Syma Khalid (Investigator), Eilish McBurnie
This project aims to use molecular dynamics techniques to understand how antimicrobial peptides, daptomycin and chlorhexidine, disrupt both gram positive and negative cell membranes on an atomic level.
Vortex Dynamics in High-Tc superconductors
Hans Fangohr (Investigator)
The dynamics of vortices in high temperature superconductors exhibits the complex and rich physics we expect from many body systems with competing interactions. Molecular Dynamics, Langevin Dynamics and Monte Carlo Computer simulations are carried out to understand this system in more detail.
People
 Kees de Groot
Kees de GrootProfessor, Electronics and Computer Science (FPAS)
 Timothy Elliott
Timothy ElliottProfessor, Medicine (FM)
 Jonathan Essex
Jonathan EssexProfessor, Chemistry (FNES)
 Hans Fangohr
Hans FangohrProfessor, Engineering Sciences (FEE)
 Paul Lewin
Paul LewinProfessor, Electronics and Computer Science (FPAS)
Nicolas GreenReader, Electronics and Computer Science (FPAS)
 Peter Horak
Peter HorakReader, Optoelectronics Research Centre
 Jorn Werner
Jorn WernerReader, Biological Sciences (FNES)
Markus BredeSenior Lecturer, Electronics and Computer Science (FPAS)
 Edward Richardson
Edward RichardsonSenior Lecturer, Engineering Sciences (FEE)
 Richard Watson
Richard WatsonSenior Lecturer, Electronics and Computer Science (FPAS)
 Ian Hawke
Ian HawkeLecturer, Mathematics (FSHS)
 Denis Kramer
Denis KramerLecturer, Engineering Sciences (FEE)
 Chris-Kriton Skylaris
Chris-Kriton SkylarisLecturer, Chemistry (FNES)
 Anatoliy Vorobev
Anatoliy VorobevLecturer, Engineering Sciences (FEE)
 Syma Khalid
Syma KhalidPrincipal Research Fellow, Chemistry (FNES)
 Philip Williamson
Philip WilliamsonSenior Research Fellow, Biological Sciences (FNES)
 Alistair Bailey
Alistair BaileyResearch Fellow, Medicine (FM)
 Jacek Dziedzic
Jacek DziedzicResearch Fellow, Chemistry (FNES)
Jia HuoResearch Fellow, Chemistry (FNES)
 Ugur Mart
Ugur MartResearch Fellow, Engineering Sciences (FEE)
Otello RoscioniResearch Fellow, Chemistry (FNES)
 Ioannis Begleris
Ioannis BeglerisPostgraduate Research Student, Engineering Sciences (FEE)
Michael BodnarchukPostgraduate Research Student, Chemistry (FNES)
Louise BoltonPostgraduate Research Student, Medicine (FM)
 Rory Brown
Rory BrownPostgraduate Research Student, Civil Engineering & the Environment (FEE)
 Christopher Cave-Ayland
Christopher Cave-AylandPostgraduate Research Student, Electronics and Computer Science (FPAS)
 Paul Chambers
Paul ChambersPostgraduate Research Student, Engineering Sciences (FEE)
 Dmitri Chernyshenko
Dmitri ChernyshenkoPostgraduate Research Student, Engineering Sciences (FEE)
 Peter Cherry
Peter CherryPostgraduate Research Student, Chemistry (FNES)
 Samuel Diserens
Samuel DiserensPostgraduate Research Student, Engineering Sciences (FEE)
Caroline DuignanPostgraduate Research Student, Biological Sciences (FNES)
 Joseph Egan
Joseph EganPostgraduate Research Student, Mathematics (FSHS)
 Robert Entwistle
Robert EntwistlePostgraduate Research Student, Engineering Sciences (FEE)
Stephen FoxPostgraduate Research Student, Chemistry (FNES)
 Ric Gillams
Ric GillamsPostgraduate Research Student, Chemistry (FNES)
 Stephen Gow
Stephen GowPostgraduate Research Student, Engineering Sciences (FEE)
 Joshua Greenhalgh
Joshua GreenhalghPostgraduate Research Student, Engineering Sciences (FEE)
 Ioannis Haldoupis
Ioannis HaldoupisPostgraduate Research Student, Chemistry (FNES)
 James Harrison
James HarrisonPostgraduate Research Student, Engineering Sciences (FEE)
 William Hurndall
William HurndallPostgraduate Research Student, Electronics and Computer Science (FPAS)
 Oliver Laslett
Oliver LaslettPostgraduate Research Student, Civil Engineering & the Environment (FEE)
Yuk LeungPostgraduate Research Student, Chemistry (FNES)
 Benjamin Lowe
Benjamin LowePostgraduate Research Student, Electronics and Computer Science (FPAS)
 David Lusher
David LusherPostgraduate Research Student, Engineering Sciences (FEE)
Jamie ParkinPostgraduate Research Student, Chemistry (FNES)
 Alvaro Perez-Diaz
Alvaro Perez-DiazPostgraduate Research Student, Engineering Sciences (FEE)
 Can Pervane
Can PervanePostgraduate Research Student, Electronics and Computer Science (FPAS)
 Lyuboslav Petrov
Lyuboslav PetrovPostgraduate Research Student, Electronics and Computer Science (FPAS)
 Chris Pittock
Chris PittockPostgraduate Research Student, Chemistry (FNES)
 Craig Rafter
Craig RafterPostgraduate Research Student, Engineering Sciences (FEE)
 Hossam Ragheb
Hossam RaghebPostgraduate Research Student, Engineering Sciences (FEE)
 Sonya Ridden
Sonya RiddenPostgraduate Research Student, Mathematics (FSHS)
 Álvaro Ruiz-Serrano
Álvaro Ruiz-SerranoPostgraduate Research Student, Chemistry (FNES)
 Kieran Selvon
Kieran SelvonPostgraduate Research Student, Engineering Sciences (FEE)
 Ashley Setter
Ashley SetterPostgraduate Research Student, Engineering Sciences (FEE)
 Valerio Vitale
Valerio VitalePostgraduate Research Student, Electronics and Computer Science (FPAS)
 Jonathon Waters
Jonathon WatersPostgraduate Research Student, Engineering Sciences (FEE)
 Thorsten Wittemeier
Thorsten WittemeierPostgraduate Research Student, Engineering Sciences (FEE)
 Emanuele Zappia
Emanuele ZappiaPostgraduate Research Student, Engineering Sciences (FEE)
 Petrina Butler
Petrina ButlerAdministrative Staff, Research and Innovation Services
 Susanne Ufermann Fangohr
Susanne Ufermann FangohrAdministrative Staff, Civil Engineering & the Environment (FEE)
 Josephine Corsi
Josephine CorsiAlumnus, University of Southampton
 Peter de_Groot
Peter de_GrootAlumnus, Physics & Astronomy (FPAS)
 Jan Kamenik
Jan KamenikAlumnus, University of Southampton
Barbara SanderAlumnus, Chemistry (FNES)
 Ian Bush
Ian BushExternal Member, NAG Ltd, Oxford
Mario OrsiExternal Member, Queen Mary University of London
Nils BerglundNone, None
Daniel HoldbrookNone, None
Eilish McBurnieNone, None
Thomas PiggotNone, None
Andrea SilvaNone, None