How far can we stretch the MARTINI?
- Research Team
- Ric Gillams
- Investigators
- Syma Khalid
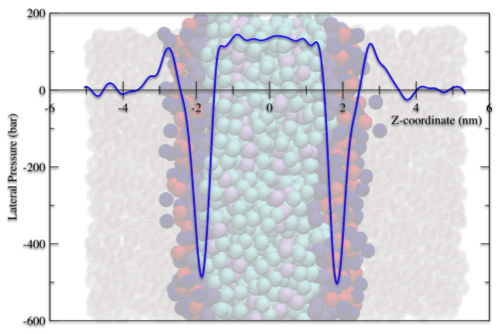
Snapshot of a DOPC lipid bilayer with lateral pressure profile overlaid.
Many biologically interesting phenomena in biomembranes are characterised by relatively large length scales (>10nm), and long time scales (multiple micro-seconds). In the foreseeable future, even with the current increase in computational power, these requirements will not be achievable with the high level of detail used in atomistic molecular dynamics simulations. To make progress in simulating biologically relevant membranes and lipids requires development of coarse-grained models that are appropriate for biological length and time scales. The MARTINI model is currently the most widely used coarse-grained model, so studies were undertaken to assess its capability to simulate correctly both the phase behaviour of bulk lipids and the mechanical properties of lipid bilayers. We have shown that this model fails to correctly reproduce the experimentally observed phase behaviour for three representative lipid species and have subsequently made modifications to increase the model’s predictive capacity. These modifications included changes to the lipid geometries and a variety of coarse-grained water models. However, we have found that even when a model is relatively successful in predicting phase behaviour, it still fails to capture the correct mechanical properties of bilayers made from these lipids.
Categories
Life sciences simulation: Biomolecular simulations, Structural biology, Systems biology
Physical Systems and Engineering simulation: Biomechanics, Elasticity, Structural dynamics
Algorithms and computational methods: FFT, Molecular Dynamics
Simulation software: Gromacs
Visualisation and data handling software: VMD, Xmgrace
Software Engineering Tools: Emacs
Programming languages and libraries: C, Python
Computational platforms: Iridis, Mac OS X
Transdisciplinary tags: Complex Systems, HPC