Hybrid quantum and classical free energy methods in computational drug optimisation
- Research Team
- Christopher Cave-Ayland
- Investigators
- Jonathan Essex, Chris-Kriton Skylaris
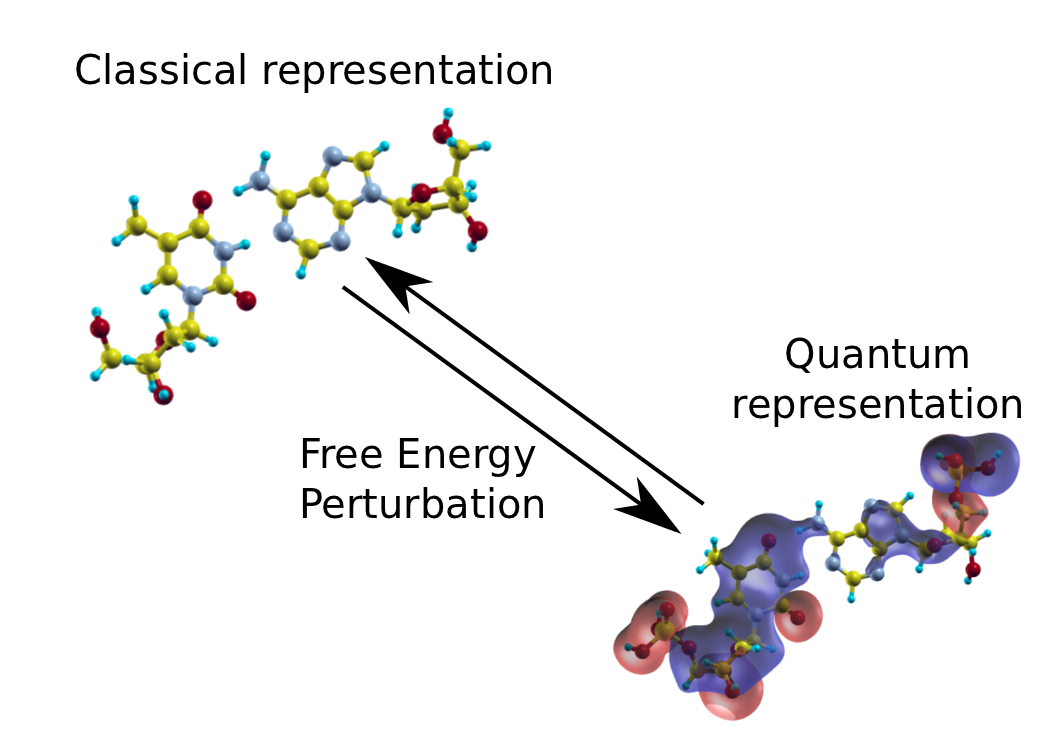
The quantum description of the same molecular structure includes explicit treatment of the electronic elements in the form of the overlaid wavefunctions. The free energy perturbation step can be used to move freely between the two different representation
Molecular systems are essentially chaotic dynamic systems that can be treated computationally to make predictions about their physical properties. The theory of thermodynamics and statistical mechanics are advanced enough to allow detailed treatments of such systems, however they depend on rigourous exploration of the configuration space of large molecules. The computational technique of Molecular Dynamics can be used to explore molecular configuration spaces but in turn depends on accurate description of the forces acting within the system. Two major ways of modelling these forces are through classical and quantum approaches. Classical approaches are highly paramaterised however relatively cheap and effectively attempt to average out the effects of the electronic components of the system. Quantum approaches on the other hand deal explicitely with the electronic structure, allowing them to capture effects that are lost in classical representations, although at a far greater computational expense. For this reason the use of Iridis is invaluable for this work as computation on a sufficient scale for even small quantum systems is impossible without a powerful and well maintained HPC environment.
My project focuses on so called hybrid classical and quantum methods, in partnership with Professor Jon Essex and Chris Skylaris from the department of chemistry. These approches attempt to gain the best of both worlds in terms of accuracy and computational tractability of both models. This effectively involves switching between the two different levels of description of the system and the presents novel practical and theoretical difficulties.
The current focus is the validation of a technique known as a free energy perturbation that can be used to transition between different descriptions of a molecular system. This is carried out through the generation of a molecular trajectory using classical methods and for a subset of configurations applying a single step free energy perturbation to reach the quantum description of the system. A number of implicit assumptions are made in the use of this technique however and by generating molecular trajectories using the quantum level of theory the intention is to perturb these to the classical state as a check for consistancy.
Categories
Life sciences simulation: Biomolecular simulations
Algorithms and computational methods: Density functional Theory, Molecular Dynamics, Molecular Mechanics, Multi-scale, Quantum Chemistry
Simulation software: AMBER, CASTEP
Visualisation and data handling software: VMD, Xmgrace
Computational platforms: Iridis, Linux
Transdisciplinary tags: Complex Systems, HPC, Scientific Computing