VMD
For queries about this topic, contact Chris-Kriton Skylaris.
View the calendar of events relating to this topic.
Projects

Antimicrobial Peptide and E. coli Membrane Interactions
Syma Khalid (Investigator), Thomas Piggot, Nils Berglund
Antimicrobial peptides (AMPs) are known to disrupt the membranes of bacterial cells such as E. coli. I work on investigating the nature of these interactions using molecular dynamics (MD) simulations.
Can we calculate the pKa of new drugs, based on their structure alone?
Chris-Kriton Skylaris (Investigator), Chris Pittock, Jacek Dziedzic
The pKa of an active compound in a pharmaceutical drug affects how it is absorbed and distributed around the human body. While there are various computational methods to predict pKa using only molecular structure data, these tend to be specialised to only one class of drug - we aim to generate a more generalised prediction method using quantum mechanics.

Centre for Doctoral Training in Next Generation Computational Modelling
Hans Fangohr, Ian Hawke, Peter Horak (Investigators), Susanne Ufermann Fangohr, Thorsten Wittemeier, Kieran Selvon, Alvaro Perez-Diaz, David Lusher, Ashley Setter, Emanuele Zappia, Hossam Ragheb, Ryan Pepper, Stephen Gow, Jan Kamenik, Paul Chambers, Robert Entwistle, Rory Brown, Joshua Greenhalgh, James Harrison, Jonathon Waters, Ioannis Begleris, Craig Rafter
The £10million Centre for Doctoral Training was launched in November 2013 and is jointly funded by EPSRC, the University of Southampton, and its partners.
The NGCM brings together world-class simulation modelling research activities from across the University of Southampton and hosts a 4-year doctoral training programme that is the first of its kind in the UK.
Dipole moment and theoretical spectroscopy: a computational approach
Chris-Kriton Skylaris (Investigator), Valerio Vitale
The present project represents a first step towards the implementation of a new technique to calculate the whole vibrational spectra of molecules in a formally exact way, which fully takes into account anharmonicity and conformational transitions, at a finite temperature, both in gas phase and in solution in a single ab initio molecular dynamics simulation.
How far can we stretch the MARTINI?
Syma Khalid (Investigator), Ric Gillams
To date, coarse-grained lipid models have generally been parameterised to ensure the correct prediction of structural properties of membranes, such as the area per lipid and the bilayer thickness. The work described here explores the extent to which coarse-grained models are able to predict correctly bulk properties of lipids (phase behaviour) as well as the mechanical properties, such as lateral pressure profiles and stored elastic stress in bilayers. Such an evaluation is crucial for understanding the predictive capabilities of coarse-grained models.
Hybrid quantum and classical free energy methods in computational drug optimisation
Jonathan Essex, Chris-Kriton Skylaris (Investigators), Christopher Cave-Ayland
This work is based around the application of thermodynamics and quantum mechanics to the field of computational drug design and optimisation. Through the application of these theories the calculation of the physical properties of drug-like molecules is possible and hence some predictive power for their pharmaceutical activity in vivo can be obtained.

Investigation into the Interfacial Physics of Field Effect Biosensors
Nicolas Green, Chris-Kriton Skylaris (Investigators), Benjamin Lowe
This interdisciplinary research aims to improve understanding of Field Effect Transistor Biosensors (Bio-FETs) and to work towards a multiscale model which can be used to better understand and predict device response.

Membrane-Protein Interactions: The Outer Membrane of Gram-Negative Bacteria
Syma Khalid (Investigator), Pin-Chia Hsu
The aim of the project is to looking for the interaction sites, which may responsible for turning on/off activity in outer membrane protein with gram-negative bacteria membrane using molecular dynamic (MD) approach.

Multi-scale simulations of bacterial outer-membrane proteins
Syma Khalid (Investigator), Jamie Parkin
Using Iridis to run multiple simulations, I aim to simulate the outer membrane proteins of Pseudomonas aeruginosa, using X-ray crystal structures of proteins only recently resolved by Bert van den Berg, University of Massachusetts. By modelling the proteins in a realistic P. aeruginosa outer membrane, I aim to gain insight into the binding of these proteins to specific substrates and their function.

Probing the oligomeric state and interaction surface of Fukutin Transmembrane Domain in lipid bilayer via Molecular Dynamics simulations
Syma Khalid, Philip Williamson (Investigators), Daniel Holdbrook, Jamie Parkin, Nils Berglund, Yuk Leung
Fukutin Transmembrane Domain (FK1TMD) is localised to the endoplasmic reticulum or Golgi Apparatus within the cell where it is believed to function as a glycosyltransferase. Its localisation within the cell is thought to be mediated by the interaction of its N-terminal transmembrane domain with the lipid bilayers surrounding these compartments, each of which possess a distinctive lipid composition. Studies have revealed that the N-terminal transmembrane domain of FK1TMD exists as dimer within dilauroylphosphatidylcholine bilayers and this interaction is driven by interactions between a characteristic TXXSS motif. Furthermore residues close to the N-terminus that have previously been shown to play a key role in the clustering of lipids are shown to play a key role in anchoring the protein in the membrane.

The application and critical assessment of protein-ligand binding affinities
Jonathan Essex (Investigator), Ioannis Haldoupis
A method that can accurately predict the binding affinity of small molecules to a protein target would be imperative to pharmaceutical development due to the time and resources that could be saved. A head-to-head comparison of such methodology, ranging from approximate methods to more rigorous methods, is performed in order to assess their accuracy and utility across a range of targets.

THE NORM MATE TRANSPORTER FROM N. GONORRHEAE: INSIGHTS INTO DRUG & ION BINDING FROM ATOMISTIC MOLECULAR DYNAMICS SIMULATIONS
Syma Khalid (Investigator), Daniel Holdbrook, Thomas Piggot, Yuk Leung
The multidrug and toxic compound extrusion (MATE) transporters extrude a wide variety of substrates out of both mammalian and bacterial cells via the electrochemical gradient of protons and cations across the membrane. Multiple atomistic simulation are performed on a MATE transporter, NorM from Neisseria gonorrheae (NorM_NG) and NorM from Vibrio cholera (NorM_VC). These simulations have allowed us to identify the nature of the drug-protein/ion-protein interactions, and secondly determine how these interactions contribute to the conformational rearrangements of the protein.

Using Molecular Dynamics to Understand the Antibacterial Mechanisms of Daptomycin & Chlorhexidine to Target the Bacterial Membrane
This project aims to use molecular dynamics techniques to understand how antimicrobial peptides, daptomycin and chlorhexidine, disrupt both gram positive and negative cell membranes on an atomic level.

Using Molecular Dynamics to Understand the Antibacterial Mechanisms of Daptomycin & Chlorhexidine to Target the Bacterial Membrane
This project aims to use molecular dynamics techniques to understand how antimicrobial peptides, daptomycin and chlorhexidine, disrupt both gram positive and negative cell membranes on an atomic level.

Using Molecular Dynamics to Understand the Antibacterial Mechanisms of Daptomycin & Chlorhexidine to Target the Bacterial Membrane
Syma Khalid (Investigator), Eilish McBurnie
This project aims to use molecular dynamics techniques to understand how antimicrobial peptides, daptomycin and chlorhexidine, disrupt both gram positive and negative cell membranes on an atomic level.

Vibrational spectroscopy from ab initio molecular dynamics
Hans Fangohr, Chris-Kriton Skylaris (Investigators), Valerio Vitale
In this project I used the Fourier transform of the time correlation function (FTTCF) formalism, that allows to compute the vibrational spectra of molecules both in gas and condensed phase, at finite temperature, in a single ab initio molecular dynamics simulation.
Water molecules in drug development: can we predict drug affinity when water molecules are involved?
Jonathan Essex (Investigator), Hannah Bruce Macdonald, Christopher Cave-Ayland
Water molecules are often found to be involved in drug-protein binding and can influence the effectiveness of a drug. We aim to aid drug design by calculating the energies involved with complexes of drugs, proteins and water molecules to predict the affinities of drug molecules.
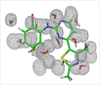
Water Molecules in Protein Binding Sites
Jonathan Essex (Investigator), Michael Bodnarchuk
Water molecules are commonplace in protein binding sites, although the true location of them can often be hard to predict from crystallographic methods. We are developing tools which enable the location and affinity of water molecules to be found.
People
 Jonathan Essex
Jonathan EssexProfessor, Chemistry (FNES)
 Hans Fangohr
Hans FangohrProfessor, Engineering Sciences (FEE)
 Nicolas Green
Nicolas GreenReader, Electronics and Computer Science (FPAS)
 Peter Horak
Peter HorakReader, Optoelectronics Research Centre
 Ian Hawke
Ian HawkeLecturer, Mathematics (FSHS)
 Chris-Kriton Skylaris
Chris-Kriton SkylarisLecturer, Chemistry (FNES)
 Syma Khalid
Syma KhalidPrincipal Research Fellow, Chemistry (FNES)
 Philip Williamson
Philip WilliamsonSenior Research Fellow, Biological Sciences (FNES)
 Alistair Bailey
Alistair BaileyResearch Fellow, Medicine (FM)
 Jacek Dziedzic
Jacek DziedzicResearch Fellow, Chemistry (FNES)
Otello RoscioniResearch Fellow, Chemistry (FNES)
Hong-Tao XueResearch Fellow, Chemistry (FNES)
 Ioannis Begleris
Ioannis BeglerisPostgraduate Research Student, Engineering Sciences (FEE)
Michael BodnarchukPostgraduate Research Student, Chemistry (FNES)
 Gabriele Boschetto
Gabriele BoschettoPostgraduate Research Student, Engineering Sciences (FEE)
 Rory Brown
Rory BrownPostgraduate Research Student, Civil Engineering & the Environment (FEE)
 Hannah Bruce Macdonald
Hannah Bruce MacdonaldPostgraduate Research Student, Chemistry (FNES)
 Christopher Cave-Ayland
Christopher Cave-AylandPostgraduate Research Student, Electronics and Computer Science (FPAS)
 Paul Chambers
Paul ChambersPostgraduate Research Student, Engineering Sciences (FEE)
 Robert Entwistle
Robert EntwistlePostgraduate Research Student, Engineering Sciences (FEE)
 Ric Gillams
Ric GillamsPostgraduate Research Student, Chemistry (FNES)
 Stephen Gow
Stephen GowPostgraduate Research Student, Engineering Sciences (FEE)
 Joshua Greenhalgh
Joshua GreenhalghPostgraduate Research Student, Engineering Sciences (FEE)
 Ioannis Haldoupis
Ioannis HaldoupisPostgraduate Research Student, Chemistry (FNES)
 James Harrison
James HarrisonPostgraduate Research Student, Engineering Sciences (FEE)
Yuk LeungPostgraduate Research Student, Chemistry (FNES)
 Benjamin Lowe
Benjamin LowePostgraduate Research Student, Electronics and Computer Science (FPAS)
 David Lusher
David LusherPostgraduate Research Student, Engineering Sciences (FEE)
Jamie ParkinPostgraduate Research Student, Chemistry (FNES)
 Alvaro Perez-Diaz
Alvaro Perez-DiazPostgraduate Research Student, Engineering Sciences (FEE)
 Can Pervane
Can PervanePostgraduate Research Student, Electronics and Computer Science (FPAS)
 Chris Pittock
Chris PittockPostgraduate Research Student, Chemistry (FNES)
 Craig Rafter
Craig RafterPostgraduate Research Student, Engineering Sciences (FEE)
 Hossam Ragheb
Hossam RaghebPostgraduate Research Student, Engineering Sciences (FEE)
 Kieran Selvon
Kieran SelvonPostgraduate Research Student, Engineering Sciences (FEE)
 Ashley Setter
Ashley SetterPostgraduate Research Student, Engineering Sciences (FEE)
 Valerio Vitale
Valerio VitalePostgraduate Research Student, Electronics and Computer Science (FPAS)
 Jonathon Waters
Jonathon WatersPostgraduate Research Student, Engineering Sciences (FEE)
 Thorsten Wittemeier
Thorsten WittemeierPostgraduate Research Student, Engineering Sciences (FEE)
 Emanuele Zappia
Emanuele ZappiaPostgraduate Research Student, Engineering Sciences (FEE)
 Susanne Ufermann Fangohr
Susanne Ufermann FangohrAdministrative Staff, Civil Engineering & the Environment (FEE)
 Jan Kamenik
Jan KamenikAlumnus, University of Southampton
 Dan Mason
Dan MasonAlumnus, University of Southampton
Nils BerglundNone, None
Daniel HoldbrookNone, None
Pin-Chia HsuNone, None
Eilish McBurnieNone, None
Thomas PiggotNone, None