Seminar 27th April 2010 3:30 p.m. B53/4025
Protein dynamics and enzyme catalysis: insights from simulations
Professor Adrian Mulholland
School of Chemistry, University of Bristol
- Categories
- Biomedical, Biomolecular simulations, Complex Systems, Density functional Theory, Materials, Molecular Dynamics, Monte Carlo, Multi-physics, Multi-scale, Systems biology
- Submitter
- Chris-Kriton Skylaris
15:30 Coffee, tea and biscuits
16:00 Start of seminar
Abstract
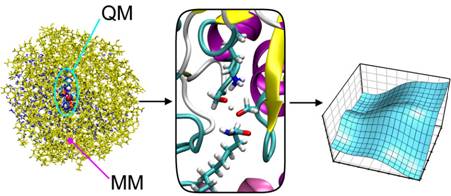
Enzymes show complex dynamics and conformational changes associated with their catalytic cycles. A central current question in understanding enzyme catalysis is what functional roles these motions may play. Biomolecular simulations have an important part to play in resolving this challenging question. Classical molecular dynamics simulations can identify functionally relevant motions on the nanosecond timescale: for example, simulations of human scavenger decapping enzyme have identified a strikingly cooperative periodic opening and closing of the protein. For modelling enzyme-catalysed reaction mechanisms, combined quantum mechanics/molecular mechanics (QM/MM) methods are a good approach. QM/MM calculations can now be carried out with highly accurate ab initio QM methods. Activation energies calculated with these methods for some enzyme reactions, such as those catalysed by chorismate mutase and p-hydroxybenzoate hydroxylase, agree very well with experiment. Such good agreement indicates that transition state theory provides a good general framework for understanding the rates of enzyme-catalysed reactions, and that dynamical effects on the rate are relatively small. Calculations based on transition state theory also give results in good agreement with experiment for enzyme reactions in which quantum tunnelling can be a significant factor: e.g. in aromatic amine dehydrogenase, calculations give results (e.g. kinetic isotope effects and barriers) in good agreement with experiment, and show that quantum tunnelling is dominant in the reaction. Simulations indicate that long-range coupled motion of the protein is not involved in ‘driving’ tunnelling. Complex conformational effects are observed in some enzymes: for example, hydrolysis of the ‘sleep inducer’ oleamide in fatty acid amide hydrolase appears to involve a minor conformation of the protein. QM/MM modelling of citrate synthase has suggested an unusual mechanism involving arginine acting as an acid; this mechanism also suggests how chemical and conformational changes may be coupled. Ligand dynamics in enzymes can also be crucial in determining reaction specificity, as shown by simulations of P450cam.
References
- ‘Computational enzymology’ (2010). R. Lonsdale, K.E. Ranaghan & A.J. Mulholland Chem. Comm., (2010) 2354-2372
- ‘Biomolecular simulation and modelling: status, progress and prospects’ (2008). M.W. van der Kamp et al. J. Royal Soc. Interface, 5, S173-S190.
- ‘Computer simulations of quantum tunnelling in enzyme-catalysed hydrogen transfer reactions’ (2010). K.E. Ranaghan & A.J. Mulholland Interdiscip. Sci. Comput. Life Sci. 2,78.
- ‘Compound I Reactivity Defines Alkene Oxidation Selectivity in Cytochrome P450cam’ (2010) R. Lonsdale, J.N. Harvey & A.J. Mulholland J. Phys. Chem. B 114, 1156-1162.