Biomolecular simulations
For queries about this topic, contact Syma Khalid.
View the calendar of events relating to this topic.
Projects

Antimicrobial Peptide and E. coli Membrane Interactions
Syma Khalid (Investigator), Thomas Piggot, Nils Berglund
Antimicrobial peptides (AMPs) are known to disrupt the membranes of bacterial cells such as E. coli. I work on investigating the nature of these interactions using molecular dynamics (MD) simulations.
Can we calculate the pKa of new drugs, based on their structure alone?
Chris-Kriton Skylaris (Investigator), Chris Pittock, Jacek Dziedzic
The pKa of an active compound in a pharmaceutical drug affects how it is absorbed and distributed around the human body. While there are various computational methods to predict pKa using only molecular structure data, these tend to be specialised to only one class of drug - we aim to generate a more generalised prediction method using quantum mechanics.
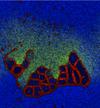
Cellular Automata Modelling of Membrane Formation and Protocell Evolution
Seth Bullock (Investigator), Stuart Bartlett
We simulated the meso-level behaviour of lipid-like particles in a range of chemical and physical environments. Self-organised protocellular structures can be shown to emerge spontaneously in systems with random, homogeneous initial conditions. Introducing an additional 'toxic' particle species and an associated set of synthesis reactions produced a new set of ecological behaviours compared to the original model of Ono and Ikegami.
Development of wide-ranging functionality in ONETEP
Chris-Kriton Skylaris (Investigator), Jacek Dziedzic
ONETEP is at the cutting edge of developments in first principles calculations. However, while the fundamental difficulties of performing accurate first-principles calculations with linear-scaling cost have been solved, only a small core of functionality is currently available in ONETEP which prevents its wide application. In this collaborative project between three Universities, the original developers of ONETEP will lead an ambitious workplan whereby the functionality of the code will be rapidly and significantly enriched.

Dual resolution simulations of lipid membrane systems
Jonathan Essex (Investigator), Kieran Selvon
This project aims to shed light on cell membrane mechanisms which are difficult to probe experimentally, in particular drug permiation across the cell membrane. If one had a full understanding of this mechanism, drugs could be designed to easily cross the membrane, or target particular embedded proteins to improve their efficacy. A reliable and robust computational method to asses a molecules permeability would be invaluable in the field of drug design, we seek to perfect such a method.
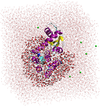
Electrostatic embedded energy calculations of proteins, using the ONETEP DFT code
Chris-Kriton Skylaris (Investigator), Stephen Fox, Chris Pittock
Calculating the energy of a biomolecule in solvent, using quantum mechanics (QM) is possible, but extremely challenging, even with linear-scaling QM methods like ONETEP. Using electrostatic embedding, a novel twist on the existing QM/MM method is used to calculate the binding energy of a small ligand to a solvated protein, increasing the accuracy and realism of our general project work.
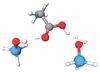
First Principles Simulation of Glycine Adsorption to Amorphous Silica
Chris-Kriton Skylaris (Investigator), Benjamin Lowe
Understanding the molecular interactions between silica and biomolecules is an important in the fields of Bionanotechnology, Biomimetic Material Science and Prebiotic Chemistry. DFT calculations were performed based on a literature study to better understand the interaction between silica and glycine.
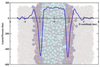
How far can we stretch the MARTINI?
Syma Khalid (Investigator), Ric Gillams
To date, coarse-grained lipid models have generally been parameterised to ensure the correct prediction of structural properties of membranes, such as the area per lipid and the bilayer thickness. The work described here explores the extent to which coarse-grained models are able to predict correctly bulk properties of lipids (phase behaviour) as well as the mechanical properties, such as lateral pressure profiles and stored elastic stress in bilayers. Such an evaluation is crucial for understanding the predictive capabilities of coarse-grained models.
Hybrid quantum and classical free energy methods in computational drug optimisation
Jonathan Essex, Chris-Kriton Skylaris (Investigators), Christopher Cave-Ayland
This work is based around the application of thermodynamics and quantum mechanics to the field of computational drug design and optimisation. Through the application of these theories the calculation of the physical properties of drug-like molecules is possible and hence some predictive power for their pharmaceutical activity in vivo can be obtained.
Immunotherapy Research: Modelling MHC Class I Complex Assembly
Timothy Elliott, Jorn Werner (Investigators), Alistair Bailey
This project uses mathematical modelling and simulation to investigate mechanisms by which our cells process and present biological information that is used by our immune system to distinguish between healthy and diseased cells.

Investigation into the Interfacial Physics of Field Effect Biosensors
Nicolas Green, Chris-Kriton Skylaris (Investigators), Benjamin Lowe
This interdisciplinary research aims to improve understanding of Field Effect Transistor Biosensors (Bio-FETs) and to work towards a multiscale model which can be used to better understand and predict device response.

Measuring biomolecules - improvements to the spectroscopic ruler
Pavlos Lagoudakis, Tom Brown (Investigators), Jan Junis Rindermann, James Richardson
The spectroscopic ruler is a technique to measure the geometry of biomolecules on the nm scale by labeling them with pairs of fluorescent markers and measuring distance dependent non-radiative energy transfer between them. The remaining uncertainty in the application of the technique originates from the unknown orientation between the optical dipole moments of the fluorescent markers, especially when the molecule undergoes thermal fluctuations in physiological conditions. Recently we introduced a simulation based method for the interpretation of the fluorescence decay dynamics of the markers that allows us to retrieve both the average orientation and the extent of directional fluctuations of the involved dipole moments.

Membrane-Protein Interactions: The Outer Membrane of Gram-Negative Bacteria
Syma Khalid (Investigator), Pin-Chia Hsu
The aim of the project is to looking for the interaction sites, which may responsible for turning on/off activity in outer membrane protein with gram-negative bacteria membrane using molecular dynamic (MD) approach.
Molecular Fragments in Inhibitor Design
Jonathan Essex (Investigator), Michael Bodnarchuk
Fragment-Based Drug Discovery (FBDD) has emerged as an important tool in the drug discovery process. Instead of screening entire drug molecules, FBDD screens molecular fragments; constituents which make up drug molecules. A computational approach to identifying fragment binding is currently being sought which also yield binding free energy estimation.

Multi-scale simulations of bacterial outer-membrane proteins
Syma Khalid (Investigator), Jamie Parkin
Using Iridis to run multiple simulations, I aim to simulate the outer membrane proteins of Pseudomonas aeruginosa, using X-ray crystal structures of proteins only recently resolved by Bert van den Berg, University of Massachusetts. By modelling the proteins in a realistic P. aeruginosa outer membrane, I aim to gain insight into the binding of these proteins to specific substrates and their function.

Probing the oligomeric state and interaction surface of Fukutin Transmembrane Domain in lipid bilayer via Molecular Dynamics simulations
Syma Khalid, Philip Williamson (Investigators), Daniel Holdbrook, Jamie Parkin, Nils Berglund, Yuk Leung
Fukutin Transmembrane Domain (FK1TMD) is localised to the endoplasmic reticulum or Golgi Apparatus within the cell where it is believed to function as a glycosyltransferase. Its localisation within the cell is thought to be mediated by the interaction of its N-terminal transmembrane domain with the lipid bilayers surrounding these compartments, each of which possess a distinctive lipid composition. Studies have revealed that the N-terminal transmembrane domain of FK1TMD exists as dimer within dilauroylphosphatidylcholine bilayers and this interaction is driven by interactions between a characteristic TXXSS motif. Furthermore residues close to the N-terminus that have previously been shown to play a key role in the clustering of lipids are shown to play a key role in anchoring the protein in the membrane.
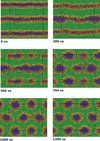
Simulation of biological systems at long length and distance scales
Jonathan Essex (Investigator), Kieran Selvon
This project aims to shed light on cell membrane mechanisms which are difficult to probe experimentally, in particular drug permiation across the cell membrane. If one had a full understanding of the mechanism, drugs could be designed to target particular embedded proteins to improve their efficacy, the viability of nano based medicines and materials could also be assessed, testing for toxicity etc.
Sustainable domain-specific software generation tools for extremely parallel particle-based simulations
Chris-Kriton Skylaris (Investigator)
A range of particle based methods (PBM) are currently used to simulate materials in chemistry, engineering, physics and biophysics. The 4 types of PBM considered directly in the proposed are molecular dynamics (MD), the ONETEP quantum mechanics-based program, discrete element modelling (DEM), and smoothed particle hydrodynamics (SPH).
The overall research objective is to develop a sustainable tool that will deliver, in the future, cutting edge research applicable to applications ranging from dam engineering to atomistic drug design.

The application and critical assessment of protein-ligand binding affinities
Jonathan Essex (Investigator), Ioannis Haldoupis
A method that can accurately predict the binding affinity of small molecules to a protein target would be imperative to pharmaceutical development due to the time and resources that could be saved. A head-to-head comparison of such methodology, ranging from approximate methods to more rigorous methods, is performed in order to assess their accuracy and utility across a range of targets.
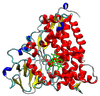
The hydrogen abstraction phase of the CYP-cyclohexene reaction, using large-scale DFT
Chris-Kriton Skylaris (Investigator), Chris Pittock, Karl Wilkinson
Studying the hydrogen-abstraction reaction between cyclohexene and the active site of cytochrome P450. This starts a series of reactions that eventually oxidise the small molecule to become either an epoxide or an alcohol.
Understanding the finer detail of this reaction can assist towards a model that will predict the breakdown of drugs in the human body.
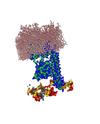
The mysteries of Braun's Lipoprotein.
Syma Khalid (Investigator), Alister Boags
Despite Braun's lipoprotein being the most common form of protein present in E.Coli bacterial membranes, it remains unstudied in the field of computational study. This is remedied by the implementation of molecular dynamics simulations to study the interactions of this lipoprotein with a structure of infinite peptidoglycan (cell wall) that has previously not been used. Shown in the project is the interactions and physical properties of the lipoprotein in it's native environment of the crowded periplasm.

THE NORM MATE TRANSPORTER FROM N. GONORRHEAE: INSIGHTS INTO DRUG & ION BINDING FROM ATOMISTIC MOLECULAR DYNAMICS SIMULATIONS
Syma Khalid (Investigator), Daniel Holdbrook, Thomas Piggot, Yuk Leung
The multidrug and toxic compound extrusion (MATE) transporters extrude a wide variety of substrates out of both mammalian and bacterial cells via the electrochemical gradient of protons and cations across the membrane. Multiple atomistic simulation are performed on a MATE transporter, NorM from Neisseria gonorrheae (NorM_NG) and NorM from Vibrio cholera (NorM_VC). These simulations have allowed us to identify the nature of the drug-protein/ion-protein interactions, and secondly determine how these interactions contribute to the conformational rearrangements of the protein.
Water molecules in drug development: can we predict drug affinity when water molecules are involved?
Jonathan Essex (Investigator), Hannah Bruce Macdonald, Christopher Cave-Ayland
Water molecules are often found to be involved in drug-protein binding and can influence the effectiveness of a drug. We aim to aid drug design by calculating the energies involved with complexes of drugs, proteins and water molecules to predict the affinities of drug molecules.
Water Molecules in Protein Binding Sites
Jonathan Essex (Investigator), Michael Bodnarchuk
Water molecules are commonplace in protein binding sites, although the true location of them can often be hard to predict from crystallographic methods. We are developing tools which enable the location and affinity of water molecules to be found.
People
 Tom Brown
Tom BrownProfessor, Chemistry (FNES)
 Seth Bullock
Seth BullockProfessor, Electronics and Computer Science (FPAS)
 Timothy Elliott
Timothy ElliottProfessor, Medicine (FM)
 Jonathan Essex
Jonathan EssexProfessor, Chemistry (FNES)
 Pavlos Lagoudakis
Pavlos LagoudakisProfessor, Physics & Astronomy (FPAS)
 Anthony Postle
Anthony PostleProfessor, Medicine (FM)
Nicolas GreenReader, Electronics and Computer Science (FPAS)
 Tiina Roose
Tiina RooseReader, Engineering Sciences (FEE)
 Jorn Werner
Jorn WernerReader, Biological Sciences (FNES)
Robert EwingSenior Lecturer, Biological Sciences (FNES)
Srinandan DasmahapatraLecturer, Electronics and Computer Science (FPAS)
 Chris-Kriton Skylaris
Chris-Kriton SkylarisLecturer, Chemistry (FNES)
 Syma Khalid
Syma KhalidPrincipal Research Fellow, Chemistry (FNES)
 Philip Williamson
Philip WilliamsonSenior Research Fellow, Biological Sciences (FNES)
 Alistair Bailey
Alistair BaileyResearch Fellow, Medicine (FM)
 Jacek Dziedzic
Jacek DziedzicResearch Fellow, Chemistry (FNES)
James RichardsonResearch Fellow, Chemistry (FNES)
Karl WilkinsonResearch Fellow, Chemistry (FNES)
 Stuart Bartlett
Stuart BartlettPostgraduate Research Student, Electronics and Computer Science (FPAS)
Michael BodnarchukPostgraduate Research Student, Chemistry (FNES)
Louise BoltonPostgraduate Research Student, Medicine (FM)
 Hannah Bruce Macdonald
Hannah Bruce MacdonaldPostgraduate Research Student, Chemistry (FNES)
 Christopher Cave-Ayland
Christopher Cave-AylandPostgraduate Research Student, Electronics and Computer Science (FPAS)
 Dmitri Chernyshenko
Dmitri ChernyshenkoPostgraduate Research Student, Engineering Sciences (FEE)
Caroline DuignanPostgraduate Research Student, Biological Sciences (FNES)
 Joseph Egan
Joseph EganPostgraduate Research Student, Mathematics (FSHS)
Stephen FoxPostgraduate Research Student, Chemistry (FNES)
 Ric Gillams
Ric GillamsPostgraduate Research Student, Chemistry (FNES)
 Ioannis Haldoupis
Ioannis HaldoupisPostgraduate Research Student, Chemistry (FNES)
 William Hurndall
William HurndallPostgraduate Research Student, Electronics and Computer Science (FPAS)
Yuk LeungPostgraduate Research Student, Chemistry (FNES)
 Benjamin Lowe
Benjamin LowePostgraduate Research Student, Electronics and Computer Science (FPAS)
Jamie ParkinPostgraduate Research Student, Chemistry (FNES)
 Can Pervane
Can PervanePostgraduate Research Student, Electronics and Computer Science (FPAS)
Maximillian PhippsPostgraduate Research Student, Chemistry (FNES)
 Chris Pittock
Chris PittockPostgraduate Research Student, Chemistry (FNES)
 Sonya Ridden
Sonya RiddenPostgraduate Research Student, Mathematics (FSHS)
 Jan Junis Rindermann
Jan Junis RindermannPostgraduate Research Student, Physics & Astronomy (FPAS)
 Kieran Selvon
Kieran SelvonPostgraduate Research Student, Engineering Sciences (FEE)
Matthew HigginsUndergraduate Research Student, Biological Sciences (FNES)
 Elena Vataga
Elena VatagaTechnical Staff, iSolutions
 Petrina Butler
Petrina ButlerAdministrative Staff, Research and Innovation Services
 Dan Mason
Dan MasonAlumnus, University of Southampton
Barbara SanderAlumnus, Chemistry (FNES)
 Shanthi Nagarajan
Shanthi NagarajanExternal Member, Korea Institute of Science and Technology
Mario OrsiExternal Member, Queen Mary University of London
Nils BerglundNone, None
Alister BoagsNone, None
Daniel HoldbrookNone, None
Pin-Chia HsuNone, None
Thomas PiggotNone, None