THE NORM MATE TRANSPORTER FROM N. GONORRHEAE: INSIGHTS INTO DRUG & ION BINDING FROM ATOMISTIC MOLECULAR DYNAMICS SIMULATIONS
- Started
- 3rd October 2011
- Ended
- 5th March 2014
- Research Team
- Daniel Holdbrook, Thomas Piggot, Yuk Leung
- Investigators
- Syma Khalid
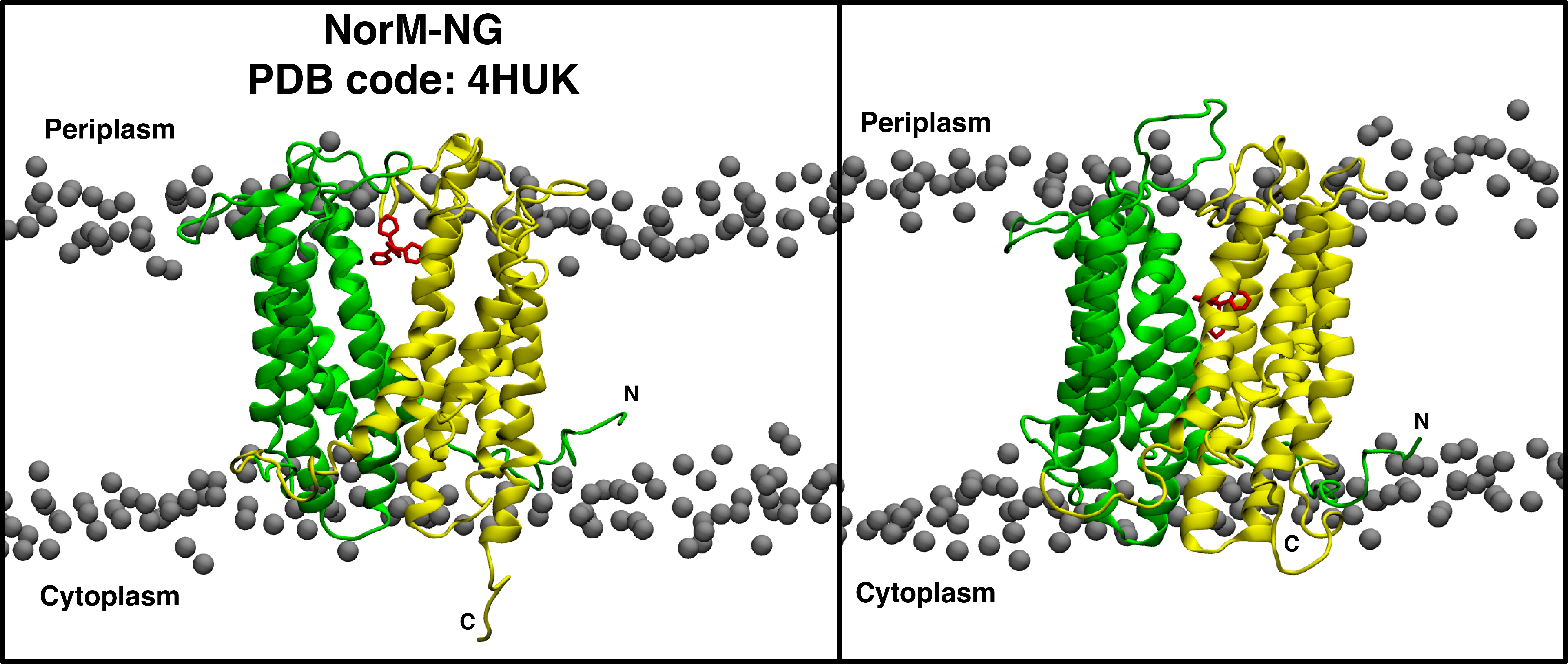
Snapshot of the simulation system before the equilibration step (left) shows the X-ray structure of the protein, NorM-NG and the drug molecule, TPP embedded in a POPC bilayer. The system after 500 ns of simulation is shown in the panel on the right. The
The Atomistic simulations of the MATE transporter (NorM_NG) identified the pathway taken by a monovalent cation to enter the cation-binding site of NorM_NG, and the key intermolecular interactions that stabilize this ion within the binding site. The presence of a bound drug molecule does not prevent movement of the ion into the binding site, but may in fact even stabilize it. In addition, the stabilization of the ion in the binding site is likely to cause the protein to proceed to the drug extrusion stage.
Finally, extra atomistic simulations were carried out on NorM_NG and NorM_VC to discover the two-ion binding state in MATE transporters, and the simulations revealed the two-ion binding state does exist in both strain of the MATE transporters.
Categories
Life sciences simulation: Biomolecular simulations
Algorithms and computational methods: Molecular Dynamics, Molecular Mechanics
Simulation software: Gromacs
Visualisation and data handling software: Gnuplot, MS Office Access, VMD, Xmgrace
Programming languages and libraries: Perl, Python, Tcl
Computational platforms: Iridis, Linux, Mac OS X
Transdisciplinary tags: Computer Science, Visualisation