Molecular Mechanics
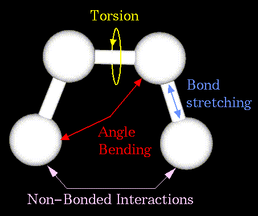
Molecular Mechanics (also known as Newtonian Mechanics) is one of the simplest methods of calculating the total energy of a molecular system, by combining the attractive energy of electrostatic and non-polar interactions, with the repulsive energy of steric geometry. Its speed of calculation makes it both a good candidate for multi-scale simulations, biomolecular dynamics and as a precursor to more advanced calculations, such as Quantum Mechanical methods.
Image taken from http://www.chem.ucla.edu/c125/NIH/MolMechanics.htm
For queries about this topic, contact Chris Pittock.
View the calendar of events relating to this topic.
Projects

Ab initio simulations of chemical reactions on platinum nanoparticles
Chris-Kriton Skylaris (Investigator), Álvaro Ruiz-Serrano, Peter Cherry
•Use first principles calculations to study the relationship between shape and size of nanoparticle and the oxygen adsorption energy.
• Investigate the effect of high oxygen coverage on the catalytic activity of the nanoparticles.
Can we calculate the pKa of new drugs, based on their structure alone?
Chris-Kriton Skylaris (Investigator), Chris Pittock, Jacek Dziedzic
The pKa of an active compound in a pharmaceutical drug affects how it is absorbed and distributed around the human body. While there are various computational methods to predict pKa using only molecular structure data, these tend to be specialised to only one class of drug - we aim to generate a more generalised prediction method using quantum mechanics.
Computational chemistry study on the interaction mechanism of imidazolium based ionic liquid lubricants with metal surface
Ugur Mart (Investigator)
We propose a fundamental research to investigate the interaction mechanism of ionic liquids (ILs) with metal surfaces, molecular structure and organization on the surface along with chemical reactions using computational chemistry methods at molecular level.
Electrostatic embedded energy calculations of proteins, using the ONETEP DFT code
Chris-Kriton Skylaris (Investigator), Stephen Fox, Chris Pittock
Calculating the energy of a biomolecule in solvent, using quantum mechanics (QM) is possible, but extremely challenging, even with linear-scaling QM methods like ONETEP. Using electrostatic embedding, a novel twist on the existing QM/MM method is used to calculate the binding energy of a small ligand to a solvated protein, increasing the accuracy and realism of our general project work.
Hybrid quantum and classical free energy methods in computational drug optimisation
Jonathan Essex, Chris-Kriton Skylaris (Investigators), Christopher Cave-Ayland
This work is based around the application of thermodynamics and quantum mechanics to the field of computational drug design and optimisation. Through the application of these theories the calculation of the physical properties of drug-like molecules is possible and hence some predictive power for their pharmaceutical activity in vivo can be obtained.

Investigation into the Interfacial Physics of Field Effect Biosensors
Nicolas Green, Chris-Kriton Skylaris (Investigators), Benjamin Lowe
This interdisciplinary research aims to improve understanding of Field Effect Transistor Biosensors (Bio-FETs) and to work towards a multiscale model which can be used to better understand and predict device response.

The application and critical assessment of protein-ligand binding affinities
Jonathan Essex (Investigator), Ioannis Haldoupis
A method that can accurately predict the binding affinity of small molecules to a protein target would be imperative to pharmaceutical development due to the time and resources that could be saved. A head-to-head comparison of such methodology, ranging from approximate methods to more rigorous methods, is performed in order to assess their accuracy and utility across a range of targets.

THE NORM MATE TRANSPORTER FROM N. GONORRHEAE: INSIGHTS INTO DRUG & ION BINDING FROM ATOMISTIC MOLECULAR DYNAMICS SIMULATIONS
Syma Khalid (Investigator), Daniel Holdbrook, Thomas Piggot, Yuk Leung
The multidrug and toxic compound extrusion (MATE) transporters extrude a wide variety of substrates out of both mammalian and bacterial cells via the electrochemical gradient of protons and cations across the membrane. Multiple atomistic simulation are performed on a MATE transporter, NorM from Neisseria gonorrheae (NorM_NG) and NorM from Vibrio cholera (NorM_VC). These simulations have allowed us to identify the nature of the drug-protein/ion-protein interactions, and secondly determine how these interactions contribute to the conformational rearrangements of the protein.
Water Molecules in Protein Binding Sites
Jonathan Essex (Investigator), Michael Bodnarchuk
Water molecules are commonplace in protein binding sites, although the true location of them can often be hard to predict from crystallographic methods. We are developing tools which enable the location and affinity of water molecules to be found.
People
 Graeme Day
Graeme DayProfessor, Chemistry (FNES)
 Jonathan Essex
Jonathan EssexProfessor, Chemistry (FNES)
 Nicolas Green
Nicolas GreenReader, Electronics and Computer Science (FPAS)
 Denis Kramer
Denis KramerLecturer, Engineering Sciences (FEE)
 Chris-Kriton Skylaris
Chris-Kriton SkylarisLecturer, Chemistry (FNES)
 Syma Khalid
Syma KhalidPrincipal Research Fellow, Chemistry (FNES)
 Jacek Dziedzic
Jacek DziedzicResearch Fellow, Chemistry (FNES)
 Ugur Mart
Ugur MartResearch Fellow, Engineering Sciences (FEE)
Otello RoscioniResearch Fellow, Chemistry (FNES)
Michael BodnarchukPostgraduate Research Student, Chemistry (FNES)
 Christopher Cave-Ayland
Christopher Cave-AylandPostgraduate Research Student, Electronics and Computer Science (FPAS)
 Peter Cherry
Peter CherryPostgraduate Research Student, Chemistry (FNES)
Caroline DuignanPostgraduate Research Student, Biological Sciences (FNES)
 Joseph Egan
Joseph EganPostgraduate Research Student, Mathematics (FSHS)
Stephen FoxPostgraduate Research Student, Chemistry (FNES)
 Ric Gillams
Ric GillamsPostgraduate Research Student, Chemistry (FNES)
 Ioannis Haldoupis
Ioannis HaldoupisPostgraduate Research Student, Chemistry (FNES)
Yuk LeungPostgraduate Research Student, Chemistry (FNES)
 Benjamin Lowe
Benjamin LowePostgraduate Research Student, Electronics and Computer Science (FPAS)
 Can Pervane
Can PervanePostgraduate Research Student, Electronics and Computer Science (FPAS)
 Lyuboslav Petrov
Lyuboslav PetrovPostgraduate Research Student, Electronics and Computer Science (FPAS)
Maximillian PhippsPostgraduate Research Student, Chemistry (FNES)
 Chris Pittock
Chris PittockPostgraduate Research Student, Chemistry (FNES)
 Álvaro Ruiz-Serrano
Álvaro Ruiz-SerranoPostgraduate Research Student, Chemistry (FNES)
Daniel HoldbrookNone, None
Thomas PiggotNone, None