AMBER
Amber, an acronym for Assisted Model Building with Energy Refinement, is a family of force fields for molecular dynamics of biomolecules originally developed by the late Peter Kollman's group at the University of California, San Francisco. AMBER is also the name for the molecular dynamics software package that simulates these force fields. It is maintained by an active collaboration between David Case at Rutgers University, Tom Cheatham at the University of Utah, Tom Darden at NIEHS, Ken Merz at Florida, Carlos Simmerling at Stony Brook University, Ray Luo at UC Irvine, and Junmei Wang at Encysive Pharmaceuticals. (Read more on Wikipedia).
For queries about this topic, contact Hans Fangohr.
View the calendar of events relating to this topic.
Projects
Can we calculate the pKa of new drugs, based on their structure alone?
Chris-Kriton Skylaris (Investigator), Chris Pittock, Jacek Dziedzic
The pKa of an active compound in a pharmaceutical drug affects how it is absorbed and distributed around the human body. While there are various computational methods to predict pKa using only molecular structure data, these tend to be specialised to only one class of drug - we aim to generate a more generalised prediction method using quantum mechanics.

Centre for Doctoral Training in Next Generation Computational Modelling
Hans Fangohr, Ian Hawke, Peter Horak (Investigators), Susanne Ufermann Fangohr, Thorsten Wittemeier, Ashley Setter, Kieran Selvon, Hossam Ragheb, Craig Rafter, Alvaro Perez-Diaz, Ryan Pepper, David Lusher, Stephen Gow, James Harrison, Paul Chambers, Jan Kamenik, Ioannis Begleris, Robert Entwistle, Jonathon Waters, Rory Brown, Joshua Greenhalgh, Emanuele Zappia
The £10million Centre for Doctoral Training was launched in November 2013 and is jointly funded by EPSRC, the University of Southampton, and its partners.
The NGCM brings together world-class simulation modelling research activities from across the University of Southampton and hosts a 4-year doctoral training programme that is the first of its kind in the UK.
Electrostatic embedded energy calculations of proteins, using the ONETEP DFT code
Chris-Kriton Skylaris (Investigator), Stephen Fox, Chris Pittock
Calculating the energy of a biomolecule in solvent, using quantum mechanics (QM) is possible, but extremely challenging, even with linear-scaling QM methods like ONETEP. Using electrostatic embedding, a novel twist on the existing QM/MM method is used to calculate the binding energy of a small ligand to a solvated protein, increasing the accuracy and realism of our general project work.
Hybrid quantum and classical free energy methods in computational drug optimisation
Jonathan Essex, Chris-Kriton Skylaris (Investigators), Christopher Cave-Ayland
This work is based around the application of thermodynamics and quantum mechanics to the field of computational drug design and optimisation. Through the application of these theories the calculation of the physical properties of drug-like molecules is possible and hence some predictive power for their pharmaceutical activity in vivo can be obtained.
Molecular Fragments in Inhibitor Design
Jonathan Essex (Investigator), Michael Bodnarchuk
Fragment-Based Drug Discovery (FBDD) has emerged as an important tool in the drug discovery process. Instead of screening entire drug molecules, FBDD screens molecular fragments; constituents which make up drug molecules. A computational approach to identifying fragment binding is currently being sought which also yield binding free energy estimation.
Sustainable domain-specific software generation tools for extremely parallel particle-based simulations
Chris-Kriton Skylaris (Investigator)
A range of particle based methods (PBM) are currently used to simulate materials in chemistry, engineering, physics and biophysics. The 4 types of PBM considered directly in the proposed are molecular dynamics (MD), the ONETEP quantum mechanics-based program, discrete element modelling (DEM), and smoothed particle hydrodynamics (SPH).
The overall research objective is to develop a sustainable tool that will deliver, in the future, cutting edge research applicable to applications ranging from dam engineering to atomistic drug design.

The application and critical assessment of protein-ligand binding affinities
Jonathan Essex (Investigator), Ioannis Haldoupis
A method that can accurately predict the binding affinity of small molecules to a protein target would be imperative to pharmaceutical development due to the time and resources that could be saved. A head-to-head comparison of such methodology, ranging from approximate methods to more rigorous methods, is performed in order to assess their accuracy and utility across a range of targets.
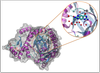
Water molecules in drug development: can we predict drug affinity when water molecules are involved?
Jonathan Essex (Investigator), Hannah Bruce Macdonald, Christopher Cave-Ayland
Water molecules are often found to be involved in drug-protein binding and can influence the effectiveness of a drug. We aim to aid drug design by calculating the energies involved with complexes of drugs, proteins and water molecules to predict the affinities of drug molecules.
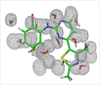
Water Molecules in Protein Binding Sites
Jonathan Essex (Investigator), Michael Bodnarchuk
Water molecules are commonplace in protein binding sites, although the true location of them can often be hard to predict from crystallographic methods. We are developing tools which enable the location and affinity of water molecules to be found.
People
 Jonathan Essex
Jonathan EssexProfessor, Chemistry (FNES)
 Hans Fangohr
Hans FangohrProfessor, Engineering Sciences (FEE)
 Nicolas Green
Nicolas GreenReader, Electronics and Computer Science (FPAS)
 Peter Horak
Peter HorakReader, Optoelectronics Research Centre
 Ian Hawke
Ian HawkeLecturer, Mathematics (FSHS)
 Chris-Kriton Skylaris
Chris-Kriton SkylarisLecturer, Chemistry (FNES)
 Jacek Dziedzic
Jacek DziedzicResearch Fellow, Chemistry (FNES)
 Ioannis Begleris
Ioannis BeglerisPostgraduate Research Student, Engineering Sciences (FEE)
Michael BodnarchukPostgraduate Research Student, Chemistry (FNES)
 Rory Brown
Rory BrownPostgraduate Research Student, Civil Engineering & the Environment (FEE)
 Hannah Bruce Macdonald
Hannah Bruce MacdonaldPostgraduate Research Student, Chemistry (FNES)
 Christopher Cave-Ayland
Christopher Cave-AylandPostgraduate Research Student, Electronics and Computer Science (FPAS)
 Paul Chambers
Paul ChambersPostgraduate Research Student, Engineering Sciences (FEE)
 Robert Entwistle
Robert EntwistlePostgraduate Research Student, Engineering Sciences (FEE)
Stephen FoxPostgraduate Research Student, Chemistry (FNES)
 Stephen Gow
Stephen GowPostgraduate Research Student, Engineering Sciences (FEE)
 Joshua Greenhalgh
Joshua GreenhalghPostgraduate Research Student, Engineering Sciences (FEE)
 Ioannis Haldoupis
Ioannis HaldoupisPostgraduate Research Student, Chemistry (FNES)
 James Harrison
James HarrisonPostgraduate Research Student, Engineering Sciences (FEE)
 David Lusher
David LusherPostgraduate Research Student, Engineering Sciences (FEE)
 Alvaro Perez-Diaz
Alvaro Perez-DiazPostgraduate Research Student, Engineering Sciences (FEE)
 Chris Pittock
Chris PittockPostgraduate Research Student, Chemistry (FNES)
 Craig Rafter
Craig RafterPostgraduate Research Student, Engineering Sciences (FEE)
 Hossam Ragheb
Hossam RaghebPostgraduate Research Student, Engineering Sciences (FEE)
 Kieran Selvon
Kieran SelvonPostgraduate Research Student, Engineering Sciences (FEE)
 Ashley Setter
Ashley SetterPostgraduate Research Student, Engineering Sciences (FEE)
 Jonathon Waters
Jonathon WatersPostgraduate Research Student, Engineering Sciences (FEE)
 Thorsten Wittemeier
Thorsten WittemeierPostgraduate Research Student, Engineering Sciences (FEE)
 Emanuele Zappia
Emanuele ZappiaPostgraduate Research Student, Engineering Sciences (FEE)
 Petrina Butler
Petrina ButlerAdministrative Staff, Research and Innovation Services
 Susanne Ufermann Fangohr
Susanne Ufermann FangohrAdministrative Staff, Civil Engineering & the Environment (FEE)
 Jan Kamenik
Jan KamenikAlumnus, University of Southampton
Barbara SanderAlumnus, Chemistry (FNES)